Protein Synthesis Inhibitors and Miscellaneous Antibacterial Agents
The antimicrobial agents discussed in this chapter may be grouped as:
• Bacteriostatic protein-synthesis inhibitors that target the ribosome, such as tetracyclines and gly-cylcyclines, chloramphenicol, macrolides and ketolides, lincosamides (clindamycin), streptogramins (quinupristin/dalfopristin), oxazolidinones (linezolid), and aminocyclitols (spectinomycin).
• Agents acting on the cell wall or cell membrane such as polymyxins, glycopeptides (vancomycin and teicoplanin), and lipopeptides (daptomycin).
• Miscellaneous compounds acting by diverse mechanisms with limited indications: bacitracin and mupirocin.
TETRACYCLINES AND GLYCYLCYCLINES
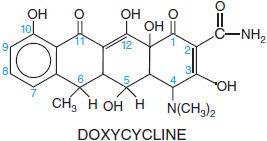
The tetracyclines are a series of derivatives of a basic 4-ring structure shown below for doxycycline. Glycylcyclines are tetracycline congers with substituents that confer broad-spectrum activity and activity against bacteria that are resistant to other antibiotics; the available glycylcycline is tigecycline (TYGACIL).
MECHANISM OF ACTION. Tetracyclines and glycylcyclines inhibit bacterial protein synthesis by binding to the 30S bacterial ribosome and preventing access of aminoacyl tRNA to the acceptor (A) site on the mRNA-ribosome complex (Figure 55–1). These drugs enter gram-negative bacteria by passive diffusion through channels formed by porins in the outer cell membrane and by active transport that pumps tetracyclines across the cytoplasmic membrane.
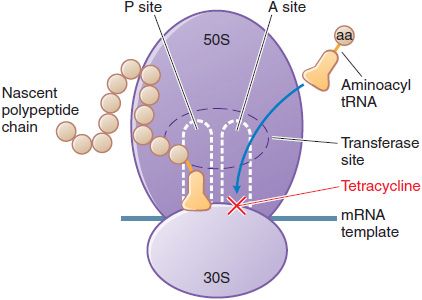
Figure 55–1 Inhibition of bacterial protein synthesis by tetracyclines. mRNA attaches to the 30S subunit of bacterial ribosomal RNA. The P (peptidyl) site of the 50S ribosomal RNA subunit contains the nascent polypeptide chain; normally, the aminoacyl tRNA charged with the next amino acid (aa) to be added moves into the A (acceptor) site, with complementary base pairing between the anticodon sequence of tRNA and the codon sequence of mRNA. Tetracyclines bind to the 30S subunit, block tRNA binding to the A site, and thereby inhibit protein synthesis.
ANTIMICROBIAL ACTIVITY. Tetracyclines are bacteriostatic antibiotics with activity against a wide range of aerobic and anaerobic gram-positive and gram-negative bacteria.
Doxycycline, the most important member of the tetracyclines, is a drug of choice for sexually transmitted diseases, rickettsial infections, plague, brucellosis, tularemia, and spirochetal infections, and is also used for treatment of respiratory tract infections including atypical pneumonia pathogens, and for skin and soft-tissue infections caused by community strains of methicillin-resistant Staphylococcus aureus (MRSA), for which minocycline also is effective. Glycylcyclines have activity against bacteria that resistant to the first- and second-generation tetracyclines.
These agents are effective against some microorganisms, such as Rickettsia, Coxiella burnetii, Mycoplasma pneumoniae, Chlamydia spp., Legionella spp., Ureaplasma, some atypical mycobacteria, and Plasmodium spp., that are resistant to cell-wall-active antimicrobial agents. The tetracyclines are active against many spirochetes, including Borrelia recurrentis, Borrelia burgdorferi (Lyme disease), Treponema pallidum (syphilis), and Treponema pertenue. Demeclocycline, tetracycline, minocycline, and doxycycline are available in the U.S. for systemic use. Resistance of a bacterial strain to any 1 member of the class may or may not result in cross-resistance to other tetracyclines. Tigecycline is generally active against organisms that are susceptible to tetracyclines as well as those with acquired resistance to tetracyclines.
Tetracyclines intrinsically are more active against gram-positive than gram-negative microorganisms, but acquired resistance is common. Recent data from the U.S. on the activity of tetracycline and other agents are displayed in Table 55–1. Bacillus anthracis and Listeria monocytogenes are susceptible. Doxycycline and minocycline can be active against some tetracycline-resistant isolates. Haemophilus influenzae is generally susceptible, but many Enterobacteriaceae have acquired resistance. Although all strains of Pseudomonas aeruginosa are resistant, 90% of strains of Burkholderia pseudomallei (the cause of melioidosis) are sensitive. Most strains of Brucella also are susceptible. Tetracyclines remain useful for infections caused by Haemophilus ducreyi (chancroid), Vibrio cholerae, and V. vulnificus, and inhibit the growth of Legionella pneumophila, Campylobacter jejuni, Helicobacter pylori, Yersinia pestis, Yersinia enterocolitica, Francisella tularensis, and Pasteurella multocida. The tetracyclines are active against many anaerobic and facultative microorganisms. Tetracycline is a drug of choice for treating actinomycosis.
Table 55–1
Activity of Selected Antimicrobials Against Key Gram-Positive Pathogens
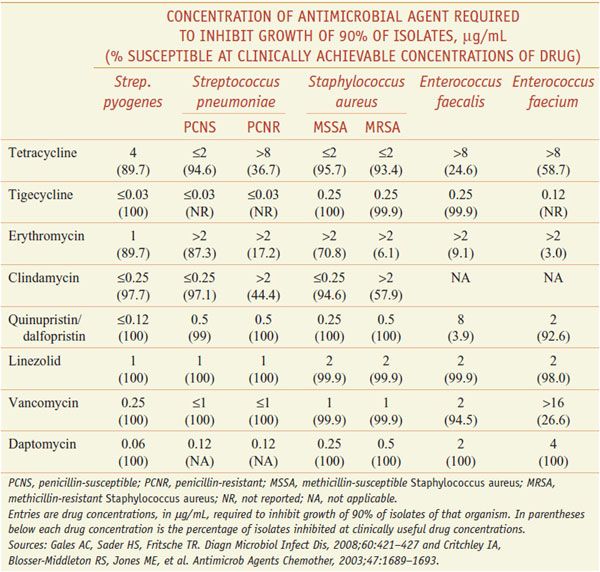
In general, tigecycline is equally or more active in vitro against bacteria than the tetracyclines, including against tetracycline-resistant organisms, especially gram-negative organisms. There are a few exceptions where other tetracyclines may be more active against certain organisms, such as Stenotrophomonas and Ureaplasma.
RESISTANCE TO TETRACYCLINES AND GLYCYLCYCLINES. Resistance is primarily plasmid mediated and often inducible. The 3 main resistance mechanisms are:
• Decreased accumulation of tetracycline as a result of either decreased antibiotic influx or acquisition of an energy-dependent efflux pathway
• Production of a ribosomal protection protein that displaces tetracycline from its target
• Enzymatic inactivation of tetracyclines
Cross-resistance, or lack thereof, among tetracyclines depends on which mechanism is operative. Tetracycline resistance due to a ribosomal protection mechanism (tetM) produces cross-resistance to doxycycline and minocycline because the target site protected is the same for all tetracyclines. The glycylamido moiety characteristic of tigecycline reduces its affinity for most efflux pumps, restoring activity against many organisms displaying tetracycline resistance due to this mechanism. Binding of glycylcyclines to ribosomes is also enhanced, improving activity against organisms that harbor ribosomal protection proteins that confer resistance to other tetracyclines.
ADME
Oral absorption of most tetracyclines is incomplete. The percentage of unabsorbed drug rises as the dose increases. Tigecycline is available only for parenteral administration. Concurrent ingestion of divalent and trivalent cations (e.g., Ca2+, Mg2+, Al3+, Fe2+/3+, and Zn2+) impairs absorption. Thus, dairy products, antacids, aluminum hydroxide gels; Ca, Mg, and Fe or Zn salts; bismuth subsalicylate (e.g., PEPTO-BISMOL) and dietary Fe and Zn supplements can interfere with absorption of tetracyclines. After a single oral dose, the peak plasma concentration is attained in 2-4 h. These drugs have half-lives in the range of 6-12 h and frequently are administered 2 to 4 times daily. Demeclocycline also is incompletely absorbed but can be administered in lower daily dosages because its t1/2 of 16 h provides effective plasma concentrations for 24-48 h.
Oral doses of doxycycline and minocycline are well absorbed (90-100%) and have half-lives of 16-18 h; they can be administered less frequently and at lower doses than tetracycline or demeclocycline. Plasma concentrations are equivalent whether doxycycline is given orally or parenterally. Food, including dairy products, does not interfere with absorption of doxycycline and minocycline.
Tetracyclines distribute widely throughout the body, including urine and prostate. They accumulate in reticuloendothelial cells of the liver, spleen, and bone marrow, and in bone, dentine, and enamel of unerupted teeth. Tigecycline distributes rapidly and extensively into tissues, with an estimated apparent volume of 7-10 L/kg. Inflammation of the meninges is not required for the passage of tetracyclines into the cerebrospinal fluid. Tetracyclines cross the placenta and enter the fetal circulation and amniotic fluid. Relatively high concentrations are found in breast milk.
Except for doxycycline, most tetracyclines are eliminated primarily by the kidney, although they also are concentrated in the liver, excreted in bile, and partially reabsorbed via enterohepatic recirculation. Comparable amounts of tetracycline (i.e., 20-60%) are excreted in the urine within 24 h following oral or intravenous administration. Doxycycline is largely excreted unchanged both in the bile and urine, tigecycline is mostly excreted unchanged along with a small amount of glucuronidated metabolites, and minocycline is extensively metabolized by the liver before excretion. Doses of these agents do not require adjustment in patients with renal dysfunction. Specific dosage adjustment recommendations in hepatic disease are available only for tigecycline. There is some evidence for drug interactions between doxycycline and hepatic enzyme-inducing agents such as phenytoin and rifampin, but not for minocycline or tigecycline.
THERAPEUTIC USES AND DOSAGE
The tetracyclines have been used extensively to treat infectious diseases and as an additive to animal feeds to facilitate growth (a use that likely contributes to the development of bacterial resistance). The drugs remain useful as first-line therapy for infections caused by rickettsiae, mycoplasmas, and chlamydiae. The glycylcyclines have restored much of the antibacterial activity lost to the tetracyclines due to resistance and can be used for a number of infections due to gram-positive and gram-negative organisms.
The oral dose of tetracycline ranges from 1-2 g/day in adults. Children >8 years of age should receive 25-50 mg/kg daily in 4 divided doses. The low pH of tetracycline, but not doxycycline or minocycline, invariably causes phlebitis if infused into a peripheral vein. The oral or intravenous dose of doxycycline for adults is 100 mg every 12 h on the first day and then 50 mg every 12 h, 100 mg once a day, or 100 mg twice daily when severe infection is present; for children >8 years of age, the dose is 4-5 mg/kg/day in 2 divided doses the first day, then 2-2.5 mg/kg given once or twice daily. The dose of minocycline for adults is 200 mg orally or intravenously initially, followed by 100 mg every 12 h; for children, it is 4 mg/kg initially followed by 2 mg/kg every 12 h. Tigecycline is administered intravenously to adults as a 100-mg loading dose, followed by 50 mg every 12 h. For patients with severe hepatic impairment, the loading dose should be followed by a reduced maintenance dose of 25 mg every 12 h. Dosage data are not available for tigecycline in pediatrics.
Tetracyclines should not be administered intramuscularly because of local irritation and poor absorption. GI distress, nausea, and vomiting can be minimized by administration of tetracyclines with food. Generally, oral administration of tetracyclines should occur 2 h before or 2 h after coadministration with any of the agents listed. Cholestyramine and colestipol also bind orally administered tetracyclines and interfere with the absorption of the antibiotic.
Respiratory Tract Infections. Doxycycline has good activity against Streptococcus pneumoniae and H. influenzae and excellent activity against atypical pathogens such as Mycoplasma and Chlamydophilia pneumoniae. Tigecycline has been demonstrated to be effective for use as a single agent for adults hospitalized with community-acquired bacterial pneumonia.
Skin and Soft-Tissue Infections. Tigecycline is approved for the treatment of complicated skin and soft-tissue infections. Low doses of tetracycline have been used to treat acne (250 mg orally twice a day).
Intra-abdominal Infections. Resistance among Enterobacteriaceae and gram-negative anaerobes limit the utility of the tetracyclines for intra-abdominal infections. However, tigecycline possesses excellent activity against these pathogens as well as Enterococcus.
GI Infections. Therapy with the tetracyclines is often ineffective in infections caused by Shigella, Salmonella, or other Enterobacteriaceae because of drug-resistant strains. Resistance limits the usefulness of tetracyclines for travelers’ diarrhea. Doxycycline (300 mg as a single dose) is effective in reducing stool volume and eradicating V. cholerae from the stool within 48 h. Some strains of V. cholerae are resistant to tetracyclines.
Sexually Transmitted Diseases. Doxycycline no longer is recommended for gonococcal infections. Chlamydia trachomatis often is a coexistent pathogen in acute pelvic inflammatory disease. Doxycycline, 100 mg intravenously twice daily, is recommended for at least 48 h after substantial clinical improvement, followed by oral therapy at the same dosage to complete a 14-day course. Acute epididymitis is caused by infection with C. trachomatis or Neisseria gonorrhoeae in men <35 years of age. Effective regimens include a single injection of ceftriaxone (250 mg) plus doxycycline, 100 mg orally twice daily for 10 days. Sexual partners also should be treated. Doxycycline (100 mg twice daily for 21 days) is first-line therapy for treatment of lymphogranuloma venereum. Non-pregnant penicillin-allergic patients who have primary, secondary, or latent syphilis can be treated with a tetracycline regimen such as doxycycline, 100 mg orally twice daily for 2 weeks. Tetracyclines should not be used for treatment of neurosyphilis.
Rickettsial Infections. Tetracyclines are life-saving in rickettsial infections, including Rocky Mountain spotted fever, recrudescent epidemic typhus (Brill disease), murine typhus, scrub typhus, rickettsialpox, and Q fever. Clinical improvement often is evident within 24 h after initiation of therapy. Doxycycline is the drug of choice for treatment of Rocky Mountain spotted fever in adults and in children, including those <9 years of age, in whom the risk of staining of permanent teeth is outweighed by the seriousness of this potentially fatal infection.
Anthrax. Doxycycline, 100 mg every 12 h (2.2 mg/kg every 12 h for children weighing <45 kg), is indicated for prevention or treatment of anthrax. It should be used in combination with another agent when treating inhalational or GI infection. The recommended duration of therapy is 60 days for bioterrorism exposures.
Local Application. Except for local use in the eye, topical use of the tetracyclines is not recommended. Minocycline sustained-release microspheres for subgingival administration are used in dentistry.
Other Infections. Tetracyclines in combination with rifampin or streptomycin are effective for acute and chronic infections caused by Brucella melitensis, Brucella suis, and Brucella abortus. Although streptomycin is preferable, tetracyclines also are effective in tularemia. Actinomycosis, although most responsive to penicillin G, may be successfully treated with a tetracycline. Minocycline is an alternative for the treatment of nocardiosis, but a sulfonamide should be used concurrently. Yaws and relapsing fever respond favorably to the tetracyclines. Tetracyclines are useful in the acute treatment and for prophylaxis of leptospirosis (Leptospira spp.). Borrelia spp., including B. recurrentis (relapsing fever) and B. burgdorferi (Lyme disease), respond to therapy with a tetracycline. The tetracyclines have been used to treat susceptible atypical mycobacterial pathogens, including Mycobacterium marinum.
UNTOWARD EFFECTS
GI. All tetracyclines can produce GI irritation, most commonly after oral administration. Tolerability can be improved by administering these drugs with food, but tetracyclines should not be taken with dairy products or antacids. Tetracycline has been associated with esophagitis, esophageal ulcers, and pancreatitis. Pseudomembranous colitis caused by overgrowth of Clostridium difficile is a potentially life-threatening complication.
Photosensitivity. Demeclocycline, doxycycline, and other tetracyclines and glycylcyclines to a lesser extent may produce photosensitivity reactions in treated individuals exposed to sunlight.
Hepatic Toxicity. Hepatic toxicity has developed in patients with renal failure receiving ≥2 g of drug per day parenterally, but this effect also may occur when large quantities are administered orally. Pregnant women are particularly susceptible.
Renal Toxicity. Tetracyclines may aggravate azotemia in patients with renal disease because of their catabolic effects. Doxycycline, minocycline, and tigecycline have fewer renal side effects than other tetracyclines. Nephrogenic diabetes insipidus has been observed in some patients receiving demeclocycline, and this phenomenon has been exploited for the treatment of the syndrome of inappropriate secretion of antidiuretic hormone (see Chapter 25). Fanconi syndrome has been observed in patients ingesting outdated tetracycline, presumably due to toxic effects on the proximal renal tubules.
Effects on Teeth. Children treated with a tetracycline or glycylcycline may develop permanent brown discoloration of the teeth. The duration of therapy appears to be less important than the total quantity of antibiotic administered. The risk is highest when a tetracycline is given to infants before the first dentition but may develop if the drug is given between the ages of 2 months and 5 years when these teeth are being calcified. Treatment of pregnant patients with tetracyclines may produce discoloration of the teeth in their children.
Other Toxic and Irritative Effects. Tetracyclines are deposited in the skeleton during gestation and throughout childhood and may depress bone growth in premature infants. This is readily reversible if the period of exposure to the drug is short. Thrombophlebitis frequently follows intravenous administration. This irritative effect of tetracyclines has been used therapeutically in patients with malignant pleural effusions. Long-term tetracycline therapy may produce leukocytosis, atypical lymphocytes, toxic granulation of granulocytes, and thrombocytopenic purpura. Tetracyclines may cause increased intracranial pressure (pseudotumor cerebri) in young infants, even when given in the usual therapeutic doses. Patients receiving minocycline may experience vestibular toxicity, manifested by dizziness, ataxia, nausea, and vomiting. The symptoms occur soon after the initial dose and generally disappear within 24-48 h after drug cessation. Various skin reactions rarely may follow the use of any of the tetracyclines. Among the more severe allergic responses are angioedema and anaphylaxis; anaphylactoid reactions can occur even after the oral use. Other hypersensitivity reactions are burning of the eyes, cheilosis, atrophic or hypertrophic glossitis, pruritus ani or vulvae, and vaginitis. Fever of varying degrees and eosinophilia may occur when these agents are administered. Asthma also has been observed. Cross-sensitization among the various tetracyclines is common.
CHLORAMPHENICOL
Chloramphenicol can cause serious and fatal blood dyscrasias; consequently, the drug is now reserved for treatment of life-threatening infections in patients who cannot take safer alternatives because of resistance or allergies.
MECHANISM OF ACTION. Chloramphenicol inhibits protein synthesis in bacteria, and to a lesser extent, in eukaryotic cells. Chloramphenicol acts primarily by binding reversibly to the 50S ribosomal subunit (near the binding site for the macrolide antibiotics and clindamycin). The drug prevents the binding of the amino acid–containing end of the aminoacyl tRNA to the acceptor site on the 50S ribosomal subunit. The interaction between peptidyltransferase and its amino acid substrate cannot occur, and peptide bond formation is inhibited (Figure 55–2).
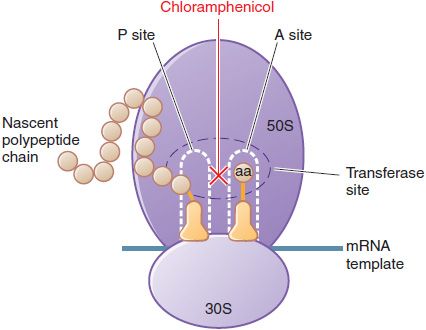
Figure 55–2 Inhibition of bacterial protein synthesis by chloramphenicol. Chloramphenicol binds to the 50S ribosomal subunit at the peptidyltransferase site, inhibiting transpeptidation. Chloramphenicol binds near the site of action of clindamycin and the macrolide antibiotics. These agents interfere with the binding of chloramphenicol and thus may interfere with each other’s actions if given concurrently. See Figure 55–1 for additional information.
Chloramphenicol also can inhibit mitochondrial protein synthesis in mammalian cells, perhaps because mitochondrial ribosomes resemble bacterial ribosomes (both are 70S); erythropoietic cells are particularly sensitive.
ANTIMICROBIAL ACTIVITY. Chloramphenicol is bacteriostatic against most species, although it may be bactericidal against H. influenzae, Neisseria meningitidis, and S. pneumoniae. Many gram-negative bacteria and most anaerobic bacteria are inhibited in vitro. Strains of S. aureus tend to be less susceptible. Chloramphenicol is active against Mycoplasma, Chlamydia, and Rickettsia. Enterobacteriaceae are variably sensitive to chloramphenicol. P. aeruginosa is resistant to even very high concentrations of chloramphenicol. Strains of V. cholerae have remained largely susceptible to chloramphenicol. Prevalent strains of Shigella and Salmonella are resistant to multiple drugs, including chloramphenicol.
RESISTANCE TO CHLORAMPHENICOL. Resistance to chloramphenicol usually is caused by a plasmid-encoded acetyltransferase that inactivates the drug. Resistance also can result from decreased permeability and from ribosomal mutation.
ADME. Chloramphenicol is absorbed rapidly from the GI tract. For parenteral use, chloramphenicol succinate is a prodrug that is hydrolyzed by esterases to chloramphenicol in vivo. Chloramphenicol succinate is rapidly cleared from plasma by the kidneys; this may reduce overall bioavailability of the drug because as much as 30% of the dose may be excreted before hydrolysis. Poor renal function in the neonate and other states of renal insufficiency result in increased plasma concentrations of chloramphenicol succinate. Decreased esterase activity has been observed in the plasma of neonates and infants, prolonging time to peak concentrations of active chloramphenicol (up to 4 h) and extending the period over which renal clearance of chloramphenicol succinate can occur.
Chloramphenicol is widely distributed in body fluids and readily reaches therapeutic concentrations in CSF. The drug actually may accumulate in the brain. Chloramphenicol is present in bile, milk, and placental fluid. It also is found in the aqueous humor after subconjunctival injection. Hepatic metabolism to the inactive glucuronide is the major route of elimination. This metabolite and chloramphenicol are excreted in the urine. Patients with impaired hepatic function have decreased metabolic clearance, and dosage should be adjusted. About 50% of chloramphenicol is bound to plasma proteins; such binding is reduced in cirrhotic patients and in neonates. Half-life is not altered significantly by renal insufficiency or hemodialysis, and dosage adjustment usually is not required. However, if the dose of chloramphenicol has been reduced because of cirrhosis, clearance by hemodialysis may be significant. This effect can be minimized by administering the drug at the end of hemodialysis. Variability in the metabolism and pharmacokinetics of chloramphenicol in neonates, infants, and children necessitates monitoring of drug concentrations in plasma.
THERAPEUTIC USES AND DOSAGE. Therapy with chloramphenicol must be limited to infections for which the benefits of the drug outweigh the risks of the potential toxicities. When other antimicrobial drugs that are equally effective and less toxic are available, they should be used instead of chloramphenicol.
Typhoid Fever. Third-generation cephalosporins and quinolones are drugs of choice for the treatment of typhoid fever. The adult dose of chloramphenicol for typhoid fever is 1 g every 6 h for 4 weeks.
Bacterial Meningitis. Chloramphenicol remains an alternative drug for the treatment of meningitis caused by H. influenzae, N. meningitidis, and S. pneumoniae in patients who have severe allergy to β-lactams and in developing countries. The total daily dose for children should be 50 mg/kg of body weight, divided into 4 equal doses given intravenously every 6 h.
Rickettsial Diseases. The tetracyclines usually are the preferred agents for the treatment of rickettsial diseases. However, in patients allergic to these drugs, in those with reduced renal function, in pregnant women, and in children <8 years of age who require prolonged or repeated courses of therapy, chloramphenicol may be the drug of choice. Rocky Mountain spotted fever, epidemic, murine, scrub, and recrudescent typhus, and Q fever respond well to chloramphenicol. For adults and children with these diseases, a dosage of 50 mg/kg/day divided into 6-h intervals is recommended. Therapy should be continued until the general condition has improved and is afebrile for 24-48 h.
UNTOWARD EFFECTS. Chloramphenicol inhibits the synthesis of proteins of the inner mitochondrial membrane, probably by inhibiting the ribosomal peptidyltransferase. Much of the toxicity observed with this drug can be attributed to these effects.
Hypersensitivity Reactions. Skin rashes may result from hypersensitivity to chloramphenicol. Fever may appear simultaneously or be the sole manifestation. Angioedema is a rare complication. Jarisch-Herxheimer reactions may occur after institution of chloramphenicol therapy for syphilis, brucellosis, and typhoid fever.
Hematological Toxicity. Chloramphenicol affects the hematopoietic system in 2 ways: a dose-related toxicity that presents as anemia, leukopenia, or thrombocytopenia, and an idiosyncratic response manifested by aplastic anemia, leading in many cases to fatal pancytopenia. Pancytopenia occurs more commonly in individuals who undergo prolonged therapy and especially in those who are exposed to the drug on more than 1 occasion. Although the incidence of the reaction is low, ~1 in ≥30,000 courses of therapy, the fatality rate is high when bone marrow aplasia is complete, and there is an increased incidence of acute leukemia in those who recover. Aplastic anemia accounts for ~70% of cases of blood dyscrasias due to chloramphenicol; hypoplastic anemia, agranulocytosis, and thrombocytopenia make up the remainder. The proposed mechanism involves conversion of the nitro group to a toxic intermediate by intestinal bacteria.
The risk of aplastic anemia does not contraindicate the use of chloramphenicol in situations in which it may be lifesaving. The drug should never be used, however, in undefined situations or in diseases readily, safely, and effectively treatable with other antimicrobial agents.
Dose-related, reversible erythroid suppression probably reflects an inhibitory action of chloramphenicol on mitochondrial protein synthesis in erythroid precursors, which in turn impairs iron incorporation into heme. Bone marrow suppression occurs regularly when plasma concentrations are ≥25 μg/mL and is observed with the use of large doses of chloramphenicol, prolonged treatment, or both. Dose-related suppression of the bone marrow may progress to fatal aplasia if treatment is continued, but most cases of bone marrow aplasia develop without prior dose-related marrow suppression.
Other Toxic and Irritative Effects. Nausea and vomiting, unpleasant taste, diarrhea, and perineal irritation may follow the oral administration of chloramphenicol. Blurring of vision and digital paresthesias may rarely occur. Tissues that have a high rate of oxygen consumption (e.g., heart, brain) may be particularly susceptible to chloramphenicol’s effects on mitochondrial enzymes.
Neonates, especially if premature, may develop a serious illness termed gray baby syndrome if exposed to excessive doses of chloramphenicol. This syndrome usually begins 2-9 days after treatment is started. Within the first 24 h, vomiting, refusal to suck, irregular and rapid respiration, abdominal distention, periods of cyanosis, and passage of loose green stools occur. Over the next 24 h, neonates turn an ashen-gray color and become flaccid and hypothermic. A similar “gray syndrome” has been reported in adults who were accidentally overdosed with the drug. Death occurs in ~40% of patients within 2 days of initial symptoms. Those who recover usually exhibit no sequelae. Two mechanisms apparently are responsible for chloramphenicol toxicity in neonates: (1) a developmental deficiency of glucuronyl transferase, the hepatic enzyme that metabolizes chloramphenicol; and (2) inadequate renal excretion of unconjugated drug. At the onset of the clinical syndrome, chloramphenicol concentrations in plasma usually exceed 100 μg/mL, and may be as low as 75 μg/mL. Children ≤2 weeks of age should receive chloramphenicol in a daily dose no larger than 25 mg/kg of body weight; after this age, full-term infants may be given daily quantities up to 50 mg/kg.
Drug Interactions. Chloramphenicol inhibits hepatic CYPs and thereby prolongs the half-lives of drugs that are metabolized by this system. Severe toxicity and death have occurred because of failure to recognize such effects. Concurrent administration of phenobarbital or rifampin, which potently induce CYPs, shortens the t1/2 of the antibiotic and may result in subtherapeutic drug concentrations.
MACROLIDES AND KETOLIDES
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


