Figure 13.1. Correlation between development phases and types of study.3 This matrix graph illustrates the relationship between the phases of development and types of study by objective that may be conducted during each clinical development of a new medicinal product. The shaded circles show the types of study most usually conducted in a certain phase of development, the open circles show certain types of study that may be conducted in that phase of development but are less usual. Each circle represents an individual study. To illustrate the development of a single study, one circle is joined by a dotted line to an inset column that depicts the elements and sequence of an individual study.
Phase 0
Phase 0 is a recent designation for exploratory, first-in-human trials conducted in accordance with the US FDA’s 2006 Guidance on Investigational New Drug (IND) studies.6 Phase 0 clinical trials are also known as human microdosing studies and are designed to speed up the development of promising drugs or imaging agents by establishing very early on whether the drug or agent behaves in human subjects as anticipated from findings in preclinical studies.
Distinctive features of Phase 0 clinical trials include the administration of single subtherapeutic dose (≤100 µg of a new chemical entity (NCE) or one-hundredth of the pharmacological dose, which ever is smaller) of the study drug to a small number of subjects (10–15) to gather preliminary data on the agent’s pharmacokinetics (how the body processes the drug) and pharmacodynamics (how the drug works in the body).
A Phase 0 study gives no data on safety or efficacy, being by definition a dose too low to cause any therapeutic effect. Drug development companies carry out Phase 0 studies to rank drug candidates in order to decide which has the best PK parameters in humans to take forward into further development. They enable ‘go/no go’ decisions that are based on relevant human models instead of relying on animal data, which can be unpredictive and vary between species. For additional information, see Chapter 11.
Phase 1
Phase 1 clinical trials are designed to determine the metabolic and pharmacologic actions of the drug in humans, the side effects associated with increasing doses (to establish a safe dose range), and, if possible, to gain early evidence of effectiveness. The ultimate goal of Phase 1 clinical trials is to obtain sufficient information about the drug’s safety, tolerability, pharmacokinetics and pharmacological effects (pharmacodynamics) to permit the design of a well-controlled, sufficiently valid Phase 2 study. Other examples of Phase 1 trials include studies on drug metabolism, structure–activity relationships and mechanisms of actions in humans, as well as studies in which investigational drugs are used as research tools to explore biological phenomena or disease processes.
The total number of subjects involved in Phase 1 clinical trials is generally in the range of 20–80. While Phase 1 clinical trials most often include healthy volunteers, there are some circumstances when patients with diseases are used as subjects, such as patients who have end-stage disease and lack other treatment options. This exception to the rule occurs most often in oncology and HIV drug trials.
Phase 1 clinical trials are often conducted in an inpatient setting, where the subject can be constantly monitored for 1–4 weeks. The subject who receives the drug is usually observed until several half-lives of the drug have passed. Phase 1 clinical trials also commonly include dose ranging (also called dose-escalation) studies so that the appropriate dose for therapeutic use can be found. The tested range of doses is usually below the dose that produces adverse events (No Observable Adverse Effect Level or NOAEL) in animals.
There are different kinds of Phase 1 clinical trials:
Phase 2
Phase 2 clinical trials include controlled clinical studies conducted to evaluate the drug’s effectiveness for a particular indication in patients with the disease or condition under study and to determine the common short-term side effects and risks associated with the drug. Some Phase 2 clinical trials are designed as case series, demonstrating a drug’s safety and activity in a selected group of patients. Other Phase 2 trials are designed as randomised clinical trials, where some patients receive the drug/device and others receive placebo/standard treatment.
Phase 2 studies are typically well-controlled, closely monitored and conducted with a relatively small number of patients, usually involving no more than several hundred patients.
When the development process for a new drug fails, it usually occurs during Phase 2 clinical trials when the drug is discovered not to have the efficacy it is presumed to have, or when it is found to have major toxic effects.
Phase 2 clinical trials are sometimes divided into the following:
Some trials combine Phase 1 and Phase 2 and test both efficacy and safety.
Following are the clinical trial design configurations usually used for Phase 2 clinical trials.
The parallel group design is the most common clinical trial design (especially for confirmatory trials, see later) in which subjects are randomly assigned to one of the two or more arms, each arm being allocated a different treatment. These treatments will include the investigational product at one or more doses and one or more control treatments, such as placebo and/or an active comparator. The subjects usually receive the assigned treatment during the entire trial. One variation of the parallel design is for each group to receive alternating (and escalating) doses of the same drug (Figure 13.2).
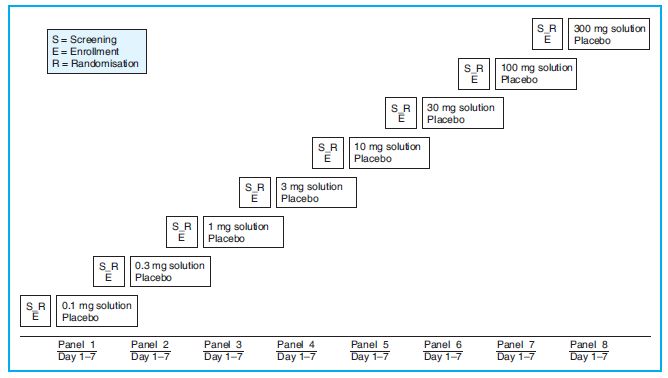
Figure 13.2. Parallel group design for clinical trial with ascending doses of study drug versus placebo. In each panel of study groups, each subject receives the study drug on Day 1, is kept under observation for 7 days and is discharged from the study on Day 7. The doses are escalated by approximately half a log.
In the simplest 2 × 2 crossover design, each subject receives each of the two treatments in randomised order in two successive treatment periods, usually separated by a washout period. Numerous variations exist, such as designs in which each subject receives a subset of n (≥2) treatments, or designs in which treatments are repeated within a subject (Table 13.1)
In a factorial design, two or more treatments are evaluated simultaneously through the use of varying combinations of the treatments. The simplest example is the 2 × 2 factorial design in which subjects are randomly allocated to one of four possible combinations of two treatments, A and B. These are as follow: A alone; B alone; both A and B; neither A nor B (i.e. placebo only). This design is used for the purpose of examining the interaction of A and B.
Factorial designs may be considered when it is possible to give the treatments together without modification (i.e., they do not interfere with each other or potentiate each other’s toxicity). This consideration is important when this design is used for examining the joint effects of A and B, in particular, if the treatments are likely to be used together.
In some cases, the 2 × 2 design may be used to make efficient use of clinical trial subjects by evaluating the efficacy of the two treatments with the same number of subjects as would be required to evaluate the efficacy of either one alone. This strategy has proved to be particularly valuable for very large mortality trials.
Table 13.2 shows the primary efficacy event rates (all-cause mortality) in patients with heart failure treated with placebo or Candesartan (CC) while also receiving low (ACEiLD) or heart-failure (ACEiHFD) dose of ACE inhibitor in the Candesartan cilexetil in Heart failure Assessment of Reduction in Mortality and morbidity (CHARM-Added) Trial.8 Although this 2 × 2 factorial analysis was not prespecified, a post-hoc analysis in FDA review showed that all-cause mortality was lowest (36.1%) in patients receiving heart-failure doses of ACE inhibitors and candesartan compared to low-dose ACE inhibitors and candesartan (39.7%), and ACE inhibitors alone at any dose plus placebo (42.1~42.2%). This finding provides support to the theory that candesartan, an angiotensin II AT-1 receptor blocker, produced a benefit on mortality on top of that produced by ACE inhibitors administered at heart-failure doses.
Another important use of the factorial design is to establish the dose–response characteristics of the simultaneous use of two treatments X and Y, especially when the efficacy of each monotherapy has been established at some dose in prior trials. A number, m, of doses of X are selected, usually including a zero dose (placebo), and a similar number, n, of doses of Y. The full design then consists of m × n treatment groups, each receiving a different combination of doses of X and Y. The resulting estimate of the response surface may then be used to help identify an appropriate combination of doses of X and Y for clinical use.4
Table 13.2. Factorial analysis of primary efficacy events in CHARM-added (Candesartan cilexetil in Heart failure Assessment of Reduction in Mortality and morbidity) Trial
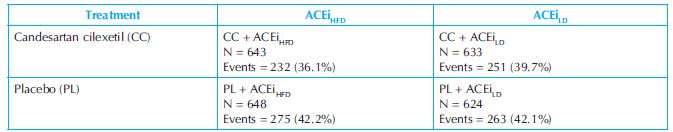
ACEi = angiotensin-converting enzyme inhibitor; HFD = heart failure dose; LD = low dose; N = number of patients.
Phase 3
Phase 3 clinical trials are performed after preliminary evidence of effectiveness has been obtained, and are intended to gather necessary additional information about effectiveness and safety for evaluating the overall benefit–risk relationship of the drug, and to provide an adequate basis for physician labelling. In Phase 3 studies, the drug is used the way it would be administered when marketed.
Phase 3 clinical trials are randomised controlled multicentre trials on commonly large outpatient groups (300–20,000 or more depending upon the disease or medical condition studied). They are confirmatory trials (pivotal trials) lasting for weeks to months and aimed at being the definitive assessment of how effective the drug is, in comparison with current ‘gold standard’ treatment, for the intended indication in the specific recipient population. Because of their size and comparatively long duration, Phase 3 clinical trials are the most expensive, time-consuming and difficult trials to design and run, especially in therapies for chronic medical conditions; for example the EUROPA (EURopean trial On reduction of cardiac events with Perindopril in stable coronary Artery disease) trial, in support of the supplementary NDA for perindopril (Aceon®) in the indication of treatment of stable coronary artery disease, treated and followed up patients for a median duration of 3.5 years.
While not required in all cases, it is typically expected that there will be at least two successful Phase 3 clinical trials, demonstrating a drug’s safety and efficacy, in order to obtain approval from the appropriate regulatory agencies (FDA [USA], Therapeutic Goods Administration [TGA; Australia], European Medicines Agency [EMEA; European Union], etc.).
Once Phase 3 trials are completed and the data analysis shows satisfactory efficacy and safety findings, the trial results are usually combined into a large document containing a comprehensive description of the methods and results of human and animal studies, manufacturing procedures, formulation details and shelf life. This collection of information makes up the ‘regulatory submission’ (in the United States, it is called an NDA). This document is provided to the appropriate regulatory authorities who will review the submission, and, depending on regulatory criteria and review findings, the US FDA will make the decision for ‘approval’, ‘approvable’ or ‘not approvable’. However, from August 2008, the FDA’s Centre for Drug Evaluation and Research has decided to replace the ‘approvable’ and ‘not approvable’ letters with a ‘complete response’ letter. ‘Complete response’ letters are already used to respond to companies that submit biologic license applications (BLAs) and they contain the following information:
Phase 4
Phase 4 clinical trials are also known as ‘Post Marketing Surveillance’ trials. Phase 4 clinical trials involve the safety surveillance (pharmacovigilance) and ongoing technical support of a drug after it receives permission to be sold. Phase 4 studies may be required by regulatory authorities. Concurrent with marketing approval, FDA may seek agreement from the sponsor to conduct certain postmarketing (Phase 4) studies to delineate additional information about the drug’s risks, benefits and optimal use. These studies could include, but would not be limited to, studying different doses or schedules of administration than were used in Phase 2 studies, use of the drug in other patient populations or other stages of the disease or use of the drug over a longer period of time [21 CFR 312.85].
Phase 4 clinical trials may be undertaken by the sponsoring company for competitive (finding a new market for the drug) or other reasons (for example, the drug may not have been tested for interactions with other drugs, or on certain population groups such as children, patients with chronic renal failure, patients with chronic liver disease, who are unlikely to subject themselves to trials). The safety surveillance is designed to detect any rare or long-term adverse effects over a much larger patient population and longer time period than was possible during the Phase 1 to Phase 3 clinical trials.
Harmful effects discovered in Phase 4 trials may result in a drug being no longer sold or restricted to certain uses: recent examples involve cerivastatin (brand names Baycol® and Lipobay®), troglitazone (Rezulin®) and rofecoxib (Vioxx®).
Designs of Confirmatory (Phase 3) Clinical Trials
Multicentre Trials
A large proportion of Phase 3 clinical trials are conducted at many sites in the same country or different countries in different regions of the world (multicentre trials).
Multicentre trials are carried out for two main reasons:
- First, a multicentre trial may present the only practical means of accruing sufficient subjects to satisfy the trial objective within a reasonable timeframe. Multicentre trials of this nature may, in principle, be carried out at any stage of clinical development. They may have several centres with a large number of subjects per centre or, in the case of a rare disease, they may have a large number of centres with very few subjects per centre.
- Second, a trial may be designed as a multicentre (and multi-investigator) trial primarily to provide a better basis to generalise findings. Recruiting subjects from a wider population and administering the medication in a broader range of clinical settings is intended to create an experimental situation that is more typical of future ‘real world’ use. In this case, the involvement of a number of investigators also gives the potential for a broader variation in clinical judgement concerning the value of the medication. Conducting a multicentre trial in a number of different countries will facilitate generalisability of results even further.
In the simplest multicentre trial, each investigator will be responsible for the subjects recruited at one hospital, so that ‘centre’ is identified uniquely by either investigator or hospital. In many trials, however, the situation is more complex. One investigator may recruit subjects from several hospitals; one investigator may represent a team of clinicians (sub-investigators) who all recruit subjects from their own clinics at one hospital or at several associated hospitals. In most instances, centres can be satisfactorily defined through the investigators.2 The aim should be to define centres to achieve homogeneity in the important factors affecting the measurements of the primary variables and the influence of the treatments.
It is important that a multicentre trial is meaningfully interpreted and extrapolated and that the manner in which the protocol is implemented is clear and similar at all centres. The sample size and power calculations depend on the assumption that the differences between the compared treatments in the centres are unbiased estimates of the same quantity. It is important to design the common protocol and to conduct the trial with this background in mind. Procedures should be standardised as completely as possible. Variation of evaluation criteria and schemes can be reduced by investigator meetings, by the training of personnel in advance of the trial and by careful monitoring during the trial.
Good design should aim to achieve the same distribution of subjects to treatments within each centre and good management should maintain this design objective. The advantage of avoiding excessive variation in the numbers of subjects per centre in a clinical trial and/or avoiding a few very small centres that participate in a clinical trial is that they reduce the differences between different weighted estimates of the treatment effect should it become necessary to take into account the heterogeneity of the treatment effect from centre to centre. (This point does not apply to trials in which ALL centres are very small and in which centre does not feature in the analysis.) Failure to take these precautions, combined with doubts about the homogeneity of the results, may, in severe cases, reduce the value of a multicentre trial to such a degree that it cannot be regarded as giving convincing evidence for the sponsor’s claims. Any rules for combining centres in the analysis should be justified and specified prospectively in the protocol where possible. Decisions concerning this approach should always be taken blind to treatment, for example, at the time of the blind statistical analysis.
Clinical Trials Designed to Show Superiority
A ‘superiority trial’ is a clinical trial which establishes efficacy of the study drug by
The advantages of the placebo-controlled clinical design are as follows:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


