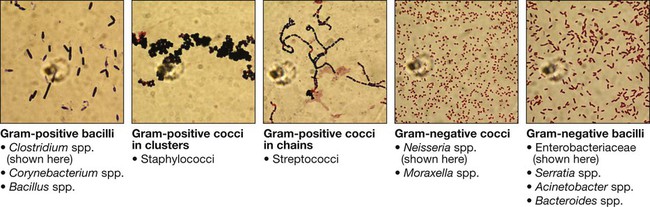
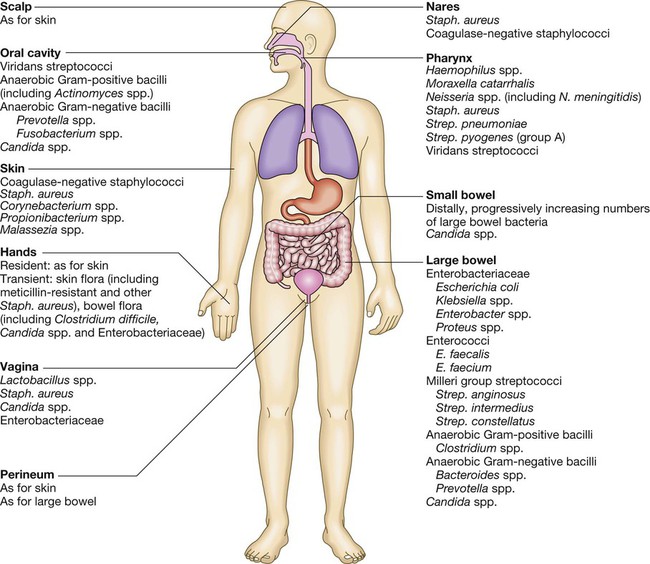
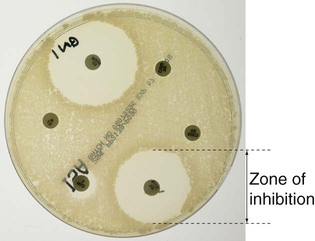
Communicable diseases are caused by organisms transmitted between hosts, whereas endogenous diseases are caused by organisms already colonising the host. Cross-infection with colonising organisms (e.g. meticillin-resistant Staphylococcus aureus, MRSA) is both communicable and endogenous. Opportunistic infections may be communicable or endogenous and arise only in individuals with impaired host defence. The chain of infection (Fig. 6.1) describes six essential elements for communicable disease transmission. Despite dramatic advances in hygiene, immunisation and antimicrobial therapy, infectious diseases are still a major cause of disease worldwide. Key challenges remain in tackling infection in resource-poor countries and in the emergence of new infectious agents and antimicrobial-resistant microorganisms. This chapter describes the biological and epidemiological principles of infectious diseases and the general approach to their prevention, diagnosis and treatment. Specific infectious diseases are described in Chapters 13–15 and many of the organ-based chapters. The concept of an infectious agent was established by Robert Koch in the 19th century (Box 6.1). Although fulfilment of ‘Koch’s postulates’ became the standard for the definition of an infectious agent, they do not apply to uncultivable organisms (e.g. Mycobacterium leprae, Tropheryma whipplei) or members of the normal human flora (e.g. Escherichia coli, Candida spp.). The following groups of infectious agents are now recognised. 1. The same organism must be present in every case of the disease 2. The organism must be isolated from the diseased host and grown in pure culture 3. The isolate must cause the disease, when inoculated into a healthy, susceptible animal 4. The organism must be re-isolated from the inoculated, diseased animal Prions are unique amongst infectious agents in that they are devoid of any nucleic acid. They appear to be transmitted by acquisition of a normal mammalian protein (prion protein, PrPC) which is in an abnormal conformation (PrPSC, containing an excess of beta-sheet protein); the abnormal protein inhibits the 26S proteasome, which can degrade misfolded proteins, leading to accumulation of the abnormally configured PrPSC protein instead of normal PrPC. The result is accumulation of protein which forms amyloid in the central nervous system, causing a transmissible spongiform encephalopathy (see Box 13.40, p. 329, and p. 1211). Viruses are incapable of independent replication, instead subverting host cellular processes to ensure synthesis of their nucleic acids and proteins. A virus that infects a bacterium is a bacteriophage (phage). Viruses contain genetic material (genome), which may be single- or double-stranded DNA or RNA. Retroviruses transcribe their RNA into DNA by reverse transcription. An antigenically unique protein coat (capsid) encloses the genome, together forming the nucleocapsid. In many viruses, the nucleocapsid is packaged within a lipid envelope. Enveloped viruses are less able to survive in the environment and are spread by respiratory, sexual or blood-borne routes, including arthropod-based transmission. Non-enveloped viruses survive better in the environment and are predominantly transmitted by faecal–oral or, less often, respiratory routes. A generic virus life cycle is shown in Figure 6.2. Prokaryotic cells are capable of synthesising their own proteins and nucleic acids, and are able to reproduce autonomously, although they lack a nucleus. The bacterial cell membrane is bounded by a peptidoglycan cell wall, which is thick (20–80 nm) in Gram-positive organisms and thin (5–10 nm) in Gram-negative ones. The Gram-negative cell wall is surrounded by an outer membrane containing lipopolysaccharide. Plasmids are rings of extra-chromosomal DNA within bacteria, which can be transferred between organisms. Bacteria may be embedded in a polysaccharide capsule, and motile bacteria are equipped with flagella. Although many prokaryotes are capable of independent existence, some (e.g. Chlamydia trachomatis, Coxiella burnetii) are obligate intracellular organisms. Bacteria that replicate in artificial culture media are classified and identified using a range of characteristics (Box 6.2), with examples in Figures 6.3 and 6.4. Eukaryotes contain functional organelles, including nuclei, mitochondria and Golgi apparatus. Eukaryotes involved in human infection include fungi, protozoa (unicellular eukaryotes with a flexible cell membrane, p. 353) and helminths (complex multicellular organisms including nematodes, trematodes and cestodes, p. 369). Fungi exist as either moulds (filamentous fungi) or yeasts. Dimorphic fungi exist in either form, depending on environmental conditions (see Fig. 13.57, p. 382). The fungal plasma membrane differs from the human cell membrane in that it contains the sterol, ergosterol. Fungi have a cell wall made up of polysaccharides, chitin and manno-proteins. In most fungi, the main structural component of the cell wall is β-1,3-D-glucan, a glucose polymer. Every human is host to an estimated 1013–1014 colonising microorganisms, which constitute the normal flora. Resident flora are able to survive and replicate at a body site, whereas transient flora are present only for short periods. Knowledge of non-sterile body sites and their normal flora is required to interpret culture results (Fig. 6.5). The normal flora contribute to endogenous disease by either excessive growth at the ‘normal’ site (overgrowth) or translocation to a sterile site. Overgrowth is exemplified by ‘blind loop’ syndrome (p. 882), dental caries and vaginal thrush, in which external factors favour overgrowth of specific components of the normal flora. Translocation results from spread along a surface or penetration of a closed barrier: for example, in urinary tract infection caused by perineal/enteric flora, and in surgical site infections, particularly of prosthetic materials, caused by skin flora such as staphylococci. Normal flora also contribute to disease by cross-infection, in which organisms that are colonising one individual cause disease when transferred to another, more susceptible, individual. • Primary pathogens cause disease in a proportion of individuals to whom they are exposed, regardless of their immunological status. • Opportunistic pathogens cause disease only in individuals whose host defences are compromised; for example, by genetic susceptibility or immunosuppressive disease or therapy. Pathogens may produce toxins, microbial molecules that cause adverse effects on host cells, either at the site of infection, or remotely following carriage through the blood stream. Endotoxin is the lipid A domain of Gram-negative bacterial outer membrane lipopolysaccharide. It is released when bacterial cells are damaged and has generalised inflammatory effects. Exotoxins are proteins released by living bacteria, which often have specific effects on target organs (Box 6.3). The harmful manifestations of infection are determined by a combination of the virulence factors of the organism and the host response to infection. Despite the obvious benefits of an intact host response, an excessive response is undesirable. Cytokines and antimicrobial factors contribute to tissue injury at the site of infection, and an excessive inflammatory response may lead to hypotension and organ dysfunction (p. 82). The contribution of the immune response to disease manifestations is exemplified by the immune reconstitution inflammatory syndrome (IRIS). This is seen, for example, in human immunodeficiency virus (HIV) infection, post-transplantation neutropenia or tuberculosis (which causes suppression of T-cell function): there is a paradoxical worsening of the clinical condition as the immune dysfunction is corrected, caused by an exuberant but dysregulated inflammatory response. Thermoregulation (p. 103) is altered in infectious disease. Microbial pyrogens or the endogenous pyrogens released during tissue necrosis stimulate specialised cells such as monocytes/macrophages to release cytokines, including interleukin (IL)-lβ, tumour necrosis factor-alpha (TNF)-α, IL-6 and interferon (IFN)-γ. Cytokine receptors in the pre-optic region of the anterior hypothalamus activate phospholipase A, releasing arachidonic acid as substrate for the cyclo-oxygenase pathway and producing prostaglandin E2 (PGE2), which in turn alters the responsiveness of thermosensitive neurons in the thermoregulatory centre. Rigors occur when the body inappropriately attempts to ‘reset’ core temperature to a higher level by stimulating skeletal muscle activity and shaking. Patients in whom a diagnosis of infectious disease is being considered are investigated with non-specific tests that reflect innate immune and acute phase responses (p. 82), and specific tests, which detect either a microorganism or the host response to the organism (Box 6.4). Careful sampling increases the likelihood of diagnosis (Box 6.5). Culture results must be interpreted in the context of the normal flora at the sampled site (see Fig. 6.5). The extent to which a microbiological test result supports or excludes a particular diagnosis depends on its statistical performance (e.g. sensitivity, specificity, positive and negative predictive value, p. 5). Sensitivity and specificity vary according to the time between infection and testing, and positive and negative predictive values depend on the prevalence of the condition in the test population. The complexity of test interpretation is illustrated in Figure 6.7 (p. 141), which shows the ‘windows of opportunity’ afforded by various testing methods. Given this complexity, effective communication between the clinician and the microbiologist is vital to ensure accurate test interpretation. • Discuss samples that may require forwarding to another laboratory or processing urgently or by an unusual method with laboratory staff before collection • Communication is the most important requirement for good microbiological sampling. If there is doubt about any aspect of sampling, it is far better to discuss it with laboratory staff beforehand than to risk diagnostic delay by inappropriate sampling or sample handling • Label sample containers and request forms according to local policies, with demographic identifiers, specimen type and time/date collected • Include clinical details on request forms • Identify samples carrying a high risk of infection (e.g. blood liable to contain a blood-borne virus) with a hazard label Whole organisms are detected by examination of biological fluids or tissue using a microscope. • Bright field microscopy (in which the test sample is interposed between the light source and the objective lens) uses stains to enhance visual contrast between the organism and its background. Examples include Gram staining of bacteria and Ziehl–Neelsen or auramine staining of acid- and alcohol-fast bacilli (AAFB) in tuberculosis. In histopathological examination of tissue samples, multiple stains are used to demonstrate not only the presence of microorganisms, but also features of disease pathology. • Dark field microscopy (in which light is scattered to make organisms appear bright on a dark background) is used, for example, to examine genital chancre fluid in suspected syphilis. • Electron microscopy may be used to examine stool and vesicle fluid to detect enteric and herpesviruses, respectively, but its use has largely been supplanted by nucleic acid detection (see below). Components of microorganisms detected for diagnostic purposes include nucleic acids, cell wall molecules, toxins and other antigens. Commonly used examples include Legionella pneumophila serogroup 1 antigen in urine and cryptococcal polysaccharide antigen in cerebrospinal fluid (CSF). Most antigen detection methods are based on in vitro binding of specific antigen/antibody and are described below (p. 141). However, other methods may be used, such as mouse bioassay for detection of Clostridium botulinum toxin or tissue culture cytotoxicity assay for C. difficile toxin. In toxin-mediated disease, detection of toxin may be of greater relevance than identification of the organism itself (e.g. stool C. difficile toxin). Specific sequences of microbial DNA and RNA are identified using a nucleic acid primer which is amplified exponentially by enzymes to generate multiple copies of the specific sequence. The most commonly used amplification method is the polymerase chain reaction (PCR; see Fig. 3.12, p. 60). Reverse transcription (RT) PCR is used to detect RNA from RNA viruses (e.g. hepatitis C virus and HIV-1). The use of fluorescent-labelled primers and probes enables ‘real-time’ detection of amplified DNA; quantification is based on the principle that the time taken to reach the detection threshold is proportional to the initial number of copies of the target nucleic acid sequence. In multiplex PCR, multiple primer pairs are used to enable detection of several different organisms in a single assay. Nucleic acid sequencing is also used to assign microorganisms to specific strains according to their genotype, which may be relevant to treatment and/or prognosis (e.g. in hepatitis C infection, p. 954). Genes that are relevant to pathogenicity (such as toxin genes) or antimicrobial resistance can also be detected. For example, detection of the mecA gene is used to screen for MRSA. • In vivo culture (in a living organism) is not used in routine diagnostic microbiology. • Ex vivo culture (tissue or cell culture) was widely used in the isolation of viruses, but has been largely supplanted by nucleic acid amplification techniques. • In vitro culture (in artificial culture media) of bacteria and fungi is used for definitive identification, to test for antimicrobial susceptibility and to subtype the organism for epidemiological purposes. Rapid microbiological diagnosis is required for blood-stream infection (BSI; Fig. 6.6). To diagnose BSI, a liquid culture medium is inoculated with freshly drawn blood, transported to the microbiology laboratory and incubated in a system that monitors it constantly for products of microbial respiration (mainly CO2), generally using fluorescence. If growth is detected, organisms are identified and sensitivity testing is performed. Traditionally, identification has been achieved by Gram stain and culture. However, MALDI-TOF (see Box 6.2) is being used increasingly, as it is rapid and inexpensive, and enables identification of organisms directly from the blood-culture medium. Organism-specific antibody detection is applied mainly to blood (Fig. 6.7). Results are typically expressed as titres: that is, the reciprocal of the highest dilution of the serum at which antibody is detectable (for example, detection at serum dilution of 1 : 64 gives a titre of 64). ‘Seroconversion’ is defined as either a change from negative to positive detection or a fourfold rise in titre between acute and convalescent serum samples. An acute sample is usually taken during the first week of disease and the convalescent sample 2–4 weeks later. Earlier diagnosis can be achieved by detection of IgM antibodies, which are produced early in infection (p. 77). A limitation of these tests is that antibody production requires a fully functional host immune system, so there may be false-negative results in immunocompromised patients. Also, other than in chronic infections and with IgM detection, antibody tests usually provide a retrospective diagnosis. The principles of the enzyme-linked immunosorbent assay (ELISA, EIA) are illustrated in Figure 6.8. These assays rely on linking an antibody with an enzyme which generates a colour change on exposure to a chromogenic substrate. Various configurations allow detection of antigens or specific subclasses of immunoglobulin (e.g. IgG, IgM, IgA). ELISA may also be adapted to detect PCR products, using immobilised oligonucleotide hybridisation probe and various detection systems. • In direct agglutination, patient serum is added to a suspension of organisms that express the test antigen. For example, in the Weil–Felix test, host antibodies to various rickettsial species cause agglutination of Proteus bacteria because they cross-react with bacterial cell surface antigens. • In indirect (passive) agglutination, specific antigen is attached to the surface of carrier particles which agglutinate when incubated with patient samples that contain specific antibodies. • In reverse passive agglutination (an antigen detection test), the carrier particle is coated with antibody rather than antigen. Immunodiffusion involves antibodies and antigen migrating through gels, with or without the assistance of electrophoresis, and forming insoluble complexes where they meet. The complexes are seen on staining as ‘precipitin bands’. Immunodiffusion is used in the diagnosis of endemic mycoses (p. 381) and some forms of aspergillosis (p. 697). Interferon-gamma release assays (IGRA) are being used increasingly to diagnose tuberculosis (p. 692). The principle of the assay is that T lymphocytes of patients infected with Mycobacterium tuberculosis (MTB) release IFN-γ when they are exposed to MTB-specific peptides. The absence of these peptides in bacille Calmette–Guérin (BCG; see Box 6.14) vaccine results in IGRA tests being more specific for the diagnosis of tuberculosis infection than the tuberculin skin test (p. 692), because the latter may be positive as a result of previous BCG vaccination. Breakpoints are determined for each antimicrobial agent from a combination of pharmacokinetic and clinical data. The relationship between in vitro antimicrobial susceptibility and clinical response is complex, as response also depends on immune status, pharmacokinetic variability (p. 21), comorbidities that may influence pharmacokinetics or pharmacodynamics, and antibiotic dosing, as well as MIC/MBC. Thus, susceptibility testing does not guarantee therapeutic success. Susceptibility testing is most often carried out by disc diffusion (Fig. 6.9). Antibiotic-impregnated filter paper discs are placed on an agar plate containing bacteria. The antibiotic diffuses through the agar, resulting in a concentration gradient centred on the disc. Bacteria are unable to grow where the antibiotic concentration exceeds the MIC, which may therefore be inferred from the size of the zone of inhibition. Susceptibility testing methods using antimicrobials diluted in liquid media are generally more accurate and reproducible, and are used for generating epidemiological data. The communicability of infectious disease means that, once a clinician has diagnosed an infectious disease, potential exposure of other patients must be considered. The patient may require treatment in isolation, or an outbreak of disease may need to be investigated in the community (Ch. 5). The approach will be specific to the microorganism involved (Chs 13–15) but the principles are outlined below. • expansion of an animal reservoir (e.g. Lyme disease from reforestation) • vector escape (e.g. airport malaria) • extension of host range (e.g. schistosomiasis from dam construction) • human migration (e.g. severe acute respiratory syndrome (SARS) coronavirus) • public health service breakdown (e.g. diphtheria in unvaccinated areas) An emerging infectious disease is one that has newly appeared in a population, or has been known for some time but is increasing in incidence or geographic range. If the disease was previously known and thought to have been controlled or eradicated, it is considered to be re-emerging. Many emerging diseases are caused by organisms which infect animals and have undergone adaptations that enable them to infect humans. This is exemplified by HIV, which is believed to have originated in higher primates in Africa. The geographical pattern of some recent emerging and re-emerging infections is shown in Figure 6.10. Infectious agents may be transmitted by one or more of the following routes: • Respiratory route: inhalation. • Faecal–oral route: ingestion of infectious material originating from faecal matter. • Sexually transmitted infections: direct contact between mucous membranes. • Blood-borne infections: direct inoculation of infected blood or body fluids. • Direct contact: very few organisms are capable of causing infection by direct contact with intact skin. Most infection by this route requires inoculation or contact with damaged skin. • Via a vector or fomite: the vector/fomite bridges the gap between the infected host or reservoir and the uninfected host. Vectors are animate, and include mosquitoes in malaria and dengue, fleas in plague and humans in MRSA. Fomites are inanimate, and include items such as door handles, water taps, ultrasound probes and so on, which are particularly associated with health care-associated infection. The likelihood of infection following transmission of an infectious agent depends on organism factors and host susceptibility. The number of organisms required to cause infection or death in 50% of the exposed population is referred to as the ID50 (infectious dose) and LD50 (lethal dose), respectively. The incubation period is the time between exposure and development of disease, and the period of infectivity is the period after exposure during which the patient is infectious to others. Knowledge of incubation periods and periods of infectivity is important in controlling the spread of disease, although for many diseases these estimates are imprecise (Boxes 6.6 and 6.7). 1From Richardson M, Elliman D, Maguire H, et al. Pediatr Infect Dis J 2001; 20:380–388. These recommendations may differ from local or national guidance. 2Transmission before 48 hrs prior to the onset of rash is rare. 3,4,5Exclude from contact with non-immune and immunocompromised people for 5 days from 3onset of rash, 4onset of parotitis or 5start of antibiotic treatment.
Principles of infectious disease
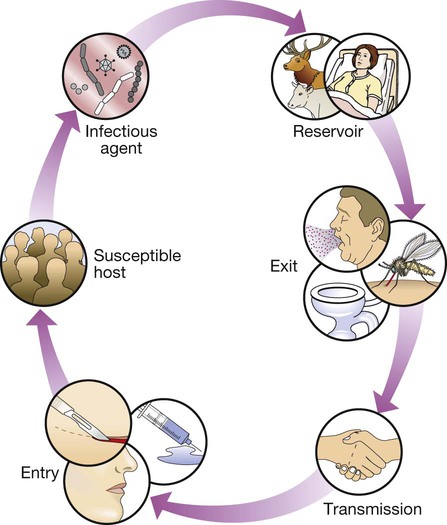
The infectious agent is the organism that causes the disease. The reservoir is the place where the population of an infectious agent is maintained. The portal of exit is the point from which the infectious agent leaves the reservoir. Transmission is the process by which the infectious agent is transferred from the reservoir to the human host, either directly or via a vector or fomite. The portal of entry is the body site that is first accessed by the infectious agent. Finally, in order for disease to ensue, the person to whom the infectious agent is transmitted must be a susceptible host.
Infectious agents
 6.1 Definition of an infectious agent – Koch’s postulates
6.1 Definition of an infectious agent – Koch’s postulates
Prions
Viruses
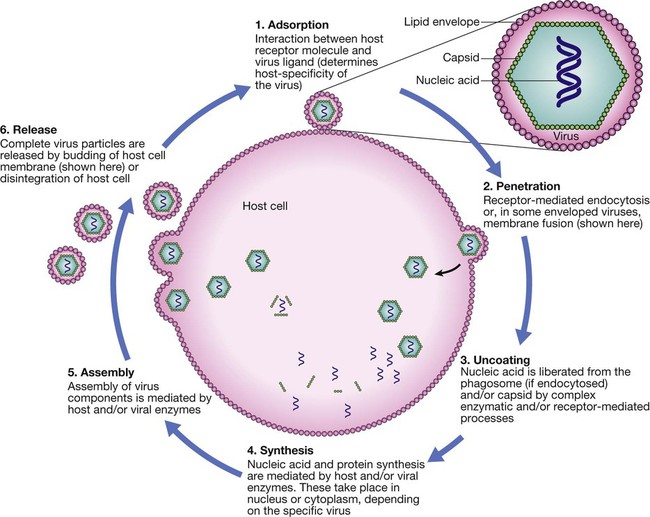
Life cycle components common to most viruses are host cell attachment and penetration, virus uncoating, nucleic acid and protein synthesis, virus assembly and release. Virus release is achieved either by budding, as illustrated, or by lysis of the cell membrane. Life cycles vary between viruses.
Prokaryotes: bacteria (including mycobacteria and actinomycetes)
 6.2 How bacteria are identified
6.2 How bacteria are identified
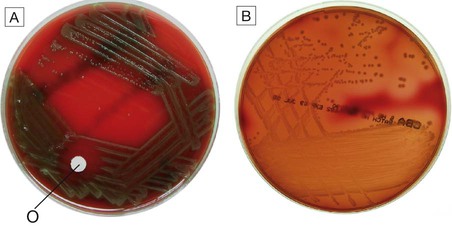
A Alpha-haemolytic streptococci. The colonies cause partial haemolysis, which imparts a green tinge to the agar. The organism shown is Strep. pneumoniae from the cerebrospinal fluid of a patient with meningitis (note also the susceptibility to optochin (O), which is another feature used to identify this organism). B Beta-haemolytic streptococci. The colonies cause complete haemolysis, which renders the agar transparent. The organism shown is Strep. pyogenes (group A β-haemolytic streptococci) from a superficial wound swab.
Eukaryotes: fungi, protozoa and helminths
Normal flora
Host–pathogen interactions
Characteristics of successful pathogens
 6.3 Exotoxin-mediated bacterial diseases
6.3 Exotoxin-mediated bacterial diseases
Disease
Organism
Antibiotic-associated diarrhoea/pseudo-membranous colitis
Clostridium difficile (p. 308)
Botulism
Clostridium botulinum (p. 1210)
Cholera
Vibrio cholerae (p. 344)
Diphtheria
Corynebacterium diphtheriae (p. 345)
Haemolytic uraemic syndrome
Escherichia coli O157 (and other strains) (p. 495)
Necrotising pneumonia
Staphylococcus aureus (p. 682)
Tetanus
Clostridium tetani (p. 1209)
Toxic shock syndrome
Staphylococcus aureus (p. 331)
Pathogenesis of infectious disease
The febrile response
Investigation of infection
 6.4 Tests used to diagnose infection
6.4 Tests used to diagnose infection
 6.5 How to provide samples for microbiological sampling
6.5 How to provide samples for microbiological sampling
Direct detection
Detection of whole organisms
Detection of components of organisms
Nucleic acid amplification tests (NAAT)
Culture
Blood culture
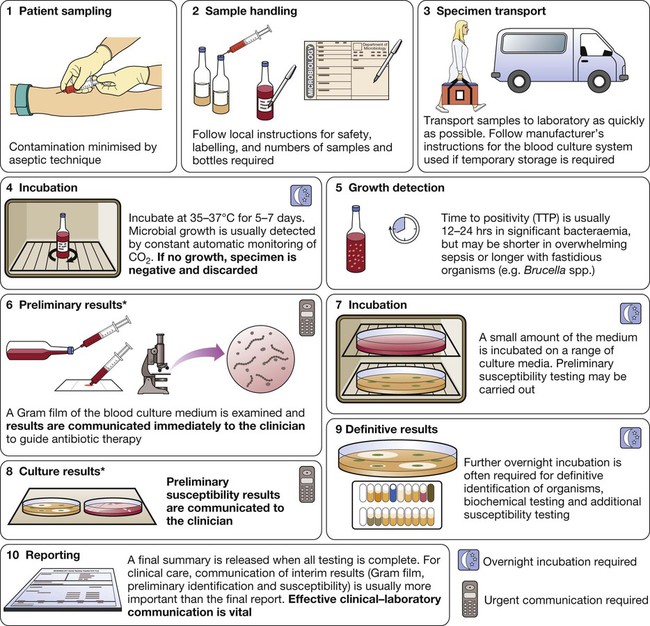
*In laboratories equipped with MALDI-TOF (p. 136), rapid definitive organism identification may be achieved at stage 6 and/or stage 8.
Specific immunological tests
Antibody detection
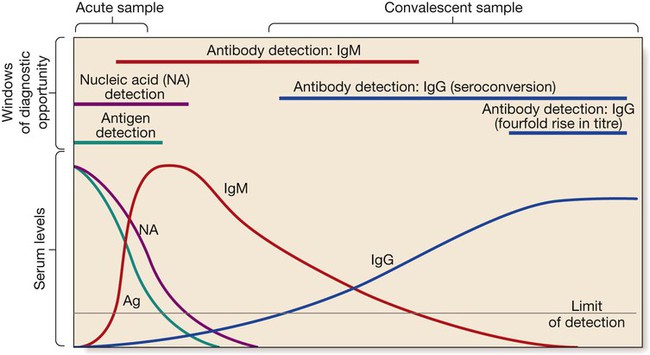
The acute sample is usually taken during the first week of illness, and the convalescent sample 2–4 weeks later. Detection limits and duration of detectability vary between tests and diseases, although in most diseases immunoglobulin (Ig) M is detectable within the first 1–2 weeks.
Enzyme-linked immunosorbent assay
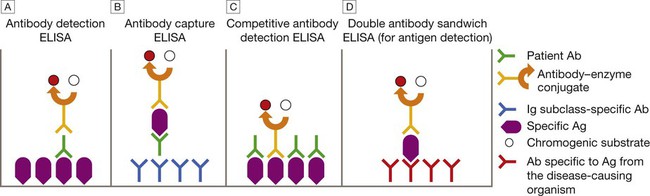
This can be configured in various ways. A Patient Ab binds to immobilised specific Ag, and is detected by addition of anti-immunoglobulin–enzyme conjugate and chromogenic substrate. B Patient Ab binds to immobilised Ig subclass-specific Ab, and is detected by addition of specific Ag, followed by antibody–enzyme conjugate and chromogenic substrate. C Patient Ab and antibody–enzyme conjugate bind to immobilised specific Ag. Magnitude of colour change reaction is inversely proportional to concentration of patient Ab. D Patient Ag binds to immobilised Ab, and is detected by addition of antibody–enzyme conjugate and chromogenic substrate. In A, the conjugate Ab is specific for human immunoglobulin. In B–D, it is specific for Ag from the disease-causing organism.
Agglutination tests
Other tests
Antibody-independent specific immunological tests
Antimicrobial susceptibility testing
Epidemiology of infection
Geographic and temporal patterns of infection
Endemic disease
Emerging and re-emerging disease
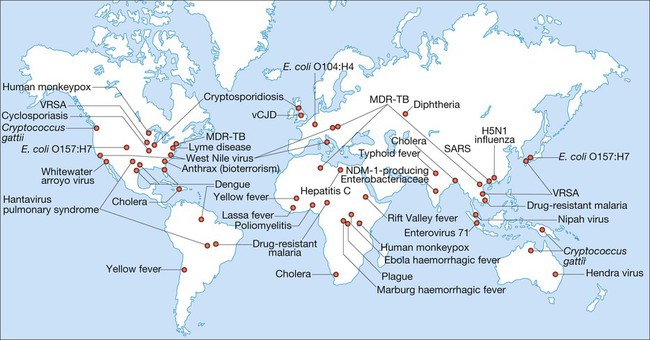
(MDR-TB = multidrug-resistant tuberculosis; SARS = severe acute respiratory syndrome; vCJD = variant Creutzfeldt–Jakob disease; VRSA = vancomycin-resistant Staph. aureus) Adapted from Samaranayake 2006 – see p. 164.
Transmission of infection
 6.6 Periods of infectivity in childhood infectious diseases1
6.6 Periods of infectivity in childhood infectious diseases1
Disease
Infectious period
Chickenpox
From 4 days before2 until 5 days after appearance of the rash3
Measles
From 1–2 days before onset of rash; duration unknown3
Mumps
Unknown4
Rubella
Unknown, but most infectious during prodromal illness3
Scarlet fever
Unknown5
Whooping cough
Unknown5,6
 6.7 Incubation periods of important infections1
6.7 Incubation periods of important infections1
Infection
Incubation period
Short incubation periods
Anthrax, cutaneous3
9 hrs to 2 wks
Anthrax, inhalational3
2 days2
Bacillary dysentery5
1–6 days
Cholera3
2 hrs to 5 days
Dengue haemorrhagic fever6
3–14 days
Diphtheria6
1–10 days
Gonorrhoea4
2–10 days
Influenza5
1–3 days
Meningococcaemia3
2–10 days
Norovirus1
1–3 days
SARS coronavirus3
2–7 days2
Scarlet fever5
2–4 days
Intermediate incubation periods
Amoebiasis6
1–4 wks
Brucellosis4
5–30 days
Chickenpox5
11–20 days
Lassa fever3
3–21 days
Malaria3
10–15 days
Measles5
6–19 days
Mumps5
15–24 days
Poliomyelitis6
3–35 days
Psittacosis4
1–4 wks
Rubella5
15–20 days
Typhoid5
5–31 days
Whooping cough5
5–21 days
Long incubation periods
Hepatitis A5
3–7 wks
Hepatitis B4
6 wks to 6 mths
Leishmaniasis, cutaneous6
Weeks to months
Leishmaniasis, visceral6
Months to years
Leprosy3
5–20 yrs
Rabies4
2–8 wks2
Trypanosoma brucei gambiense infection6
Months to years
Tuberculosis5
1–12 mths ![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree