Precursor Lymphoid Neoplasms
Diane C. Farhi
GENERAL FEATURES
Nomenclature
New precursor lymphoid neoplasms include “solid” tumors or lymphoblastic lymphoma, and “liquid” tumors or acute lymphoblastic leukemia (ALL). Lymphoblastic lymphoma may develop a “liquid” phase, with peripheral blood and bone marrow disease. ALL, in turn, cause mass lesions. Sometimes the origin is not clear, and the case should be regarded as a lymphoblastic malignancy. This chapter discusses ALL. Lymphoblastic lymphoma is discussed with malignant lymphoma.
Acute leukemia arising from lymphocytes has been variously referred to as lymphoblastic, lymphocytic, and lymphoid, with little, if any, clarification as to the meaning implied by these terms. In this chapter, the overall designation “lymphoblastic” is used to include acute leukemias arising from lymphocytes, regardless of the maturity of the immunophenotype.
Definition and Classification
ALL is a malignant clonal proliferation of lymphocytes, usually expressing an immature phenotype (1, 2, 3, 4, 5, 6, 7). It is currently classified primarily according to immunophenotypic and genetic characteristics.
The World Health Organization (WHO) classification groups of 2001 ALL with lymphoblastic lymphoma and was based primarily on immunophenotype (Table 19.1; see Appendix A for WHO 2008 classifications). The earlier French-American-British (FAB) classification was based primarily on morphologic characteristics and is no longer widely used as a primary means of risk assessment, although the blast descriptions are still very useful. At the recently introduced WHO 2008, classification is based primarily on genotype.
ALL is currently diagnosed with a peripheral blood or bone marrow blast percentage of 20% or more. Cases presenting primarily as tissue infiltrates or masses with less than 20% bone marrow blasts are usually diagnosed as lymphoblastic lymphoma.
Discussed below are subtypes of ALL, representing some of the most common, distinctive, and’or prognostically important entities. These are organized by morphology, immunophenotype, and genotype. Morphology, immunophenotype, and genotype do not correlate in many instances. It should be kept in mind that these distinctions are drawn for convenience and do not constitute a formal classification.
Etiology
ALL has been investigated for many years, with an effort made to identify etiologic risk factors (see Chapter 17 for further discussion) (8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34). Some factors are inherited and some are acquired in utero or later in life.
ALL usually occurs de novo. It may also present as a therapy-related malignancy, transformation of pre-existing lymphoma, a blast crisis of chronic myeloid leukemia or other myeloproliferative neoplasm, a component of 8p11 and other myeloproliferative neoplasms, and a recurrence of acute myeloid leukemia with phenotypic switch to acute lymphoblastic leukemia.
Considerable evidence points to the importance of intrauterine acquisition of chromosomal alterations, which may then spread to other fetuses in a multiple pregnancy or, rarely, to the mother. The presence of translocation is not equivalent to leukemia. Approximately 1% of normal newborns have a small clone of cells with t(12;21)(p13;q22), without evidence of leukemia. Likewise, patients may retain t(12;21) after successful chemotherapy for ALL, with no evidence of relapse or very late relapse of the original leukemic clone.
Clearly, in many cases acquisition of a genetic anomaly is necessary but not sufficient for the development of acute leukemia. Combinations of risk factors are common, indicating that no single risk factor may be enough to initiate a clone and produce and sustain leukemic clonal expansion.
Clinical Course
ALL occurs predominantly in children 2 to 9 years old and then shows a small but steadily rising incidence with age (35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46). The prognosis is worse in patients less than 1 year of age and in older patients.
Patients usually present with circulating blasts, cytopenias due to marrow replacement, lymphadenopathy, hepatosplenomegaly, and’or coagulopathy.
TABLE 19.1 WHO 2008 Classification of Precursor Lymphoid Neoplasms | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
Skeletal lesions at the time of diagnosis are associated with favorable prognosis. This type of ALL features prominent bone pain, low white blood cell count, and few circulating blasts.
ALL has a generally downhill course, if untreated, complicated by the sequelae of bone marrow failure. Combination chemotherapy induces remission in 80% to 90% of patients, with relapses occurring in most adult patients. Stem cell transplantation offers the best chance for lasting remission in adults.
Prognostic scores have been developed for ALL using age, disease, stage at transplantation, cytogenetics, and pretransplantation ferritin as the variables, or other criteria. In this way, ALL can be stratified into broad groups, representing low, intermediate, and high risk of mortality.
Spontaneous (or near-spontaneous) remission has rarely been reported in ALL in association with severe infection, blood transfusion, and granulocyte colony-stimulating factor therapy. It may be durable, or followed by relapse of ALL.
Aplastic Anemia: An Unusual Prodrome of Acute Lymphoblastic Leukemia
An aplastic anemia-like presentation occurs in approximately 2% of ALL, apparently initiated by viral infection in at least some cases (47, 48, 49, 50, 51, 52, 53, 54, 55, 56). Parvovirus B19 is usually implicated. The peripheral blood and bone marrow show hematopoietic hypoplasia and, in some cases, bone marrow necrosis or prominent plasmacytosis. Clonal genotypic changes typical of ALL have been demonstrated during the aplastic prodrome.
Following recovery of hematopoiesis, which usually occurs within a few days or weeks, either regression of bone marrow blasts or progression to overt ALL may occur. Interestingly, in several cases the blast karyotype showed t(9;22), or the Philadelphia chromosome.
This unusual prodrome should not be confused with acute lymphoblastic leukemia presenting with, or later developing, parvovirus B19 infection.
Peripheral Blood Findings and Blast Morphology
The peripheral blood usually shows anemia and thrombocytopenia in ALL (57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67). Red blood cell and platelet morphology is usually unremarkable. The white blood cell count and peripheral blast percentage are highly variable. In cases of pancytopenia, the peripheral blood smear should be carefully searched for blasts.
Blasts are generally round with a high nuclear: cytoplasmic ratio (Fig. 19.1). They vary in size from only slightly larger than a normal lymphocyte to over twice the diameter of a lymphocyte. It may be difficult to distinguish lymphocytes from lymphoblasts. Helpful signs include (a) the evenly dispersed chromatin of lymphoblasts, as compared to the condensed chromatin of lymphocytes, and (b) the presence of a range of blast sizes, with a spectrum ranging from small, lymphocyte-like blasts to large blasts with dispersed chromatin and scant cytoplasm. Unusually large and bizarre blasts have been reported in a case with polyploidy.
Nucleoli may be absent or single and prominent, a feature used in the FAB classification of ALL. The nuclear membrane is usually smooth but may be cleaved and convoluted, a feature initially noted in precursor T-cell ALL but later also recognized in precursor B-cell ALL.
The cytoplasmic membrane is generally smooth. A rare case of precursor T-cell ALL has been reported showing hairy projections of the cytoplasmic membrane. Blast cytoplasm is typically agranular, except for relatively rare cases of granular ALL (see below).
Other abnormalities found in the peripheral blood include eosinophilia, basophilia, and cyclic neutropenia. ALL presenting with thrombocytosis should be carefully distinguished from chronic myeloid leukemia presenting in lymphoid blast crisis.
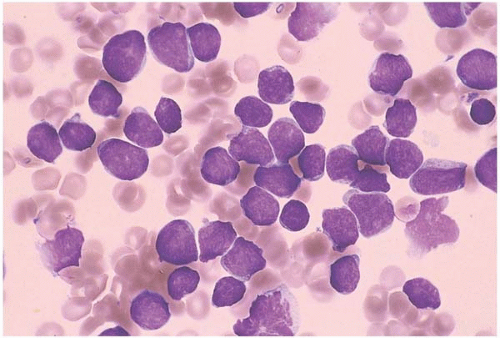 FIG. 19.1 Acute lymphoblastic leukemia (FAB L1), bone marrow aspirate. This is the most common type of childhood acute lymphoid leukemia, with numerous small, lymphocyte-like blasts admixed with larger blasts. The chromatin is fine and smooth, and the cytoplasm is nearly imperceptible. |
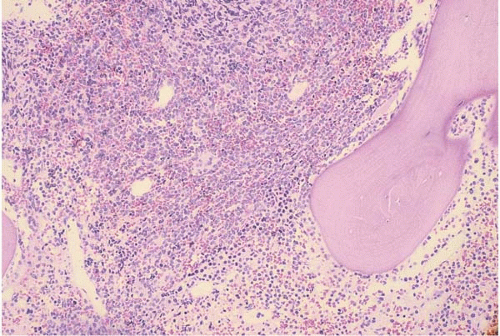 FIG. 19.2 Acute lymphoblastic leukemia (FAB L1), bone marrow biopsy. The bone marrow is replaced by a diffuse sheet infiltrate of small blasts. No residual hematopoietic tissue is seen. |
Bone Marrow Findings
Bone marrow aspirate smears show, by definition, 20% or more blasts (Figs. 19.2 and 19.3) (68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80). In practice, ALL usually presents with more than 50% blasts, and the distinction between ALL and disorders with fewer blasts is not difficult. The myeloid:erythroid ratio may be unevaluable because of the preponderance of blasts. Normal hematopoietic cells may persist but do not show myelodysplastic changes. Pseudo-Gaucher cells may be present.
Histologic sections show hypercellularity for site and age. Some cases present with hypocellularity and an aplastic anemia-like prodrome (see above). Persistence of blasts in the bone marrow after initiation of chemotherapy is a poor prognostic sign.
Other bone marrow findings include necrosis, fibrosis, osteopenia, osteolysis, and other bony abnormalities.
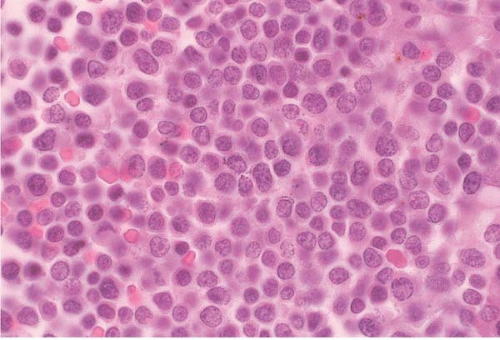 FIG. 19.3 Acute lymphoblastic leukemia (FAB L1), bone marrow biopsy. At higher power, the bone marrow is seen to be replaced by a uniform, monotonous infiltrate of small round blasts. |
ALL, like lymphoblastic lymphoma, has been reported to accompany or precede Langerhans cell histiocytosis (LCH). The lymphoblast immunophenotype, when reported, is usually precursor T-cell. In some cases, the lymphoblastic malignancy and LCH are clonally related.
Cytochemistry
Cytochemical stains have been largely supplanted by immunophenotyping for the diagnosis of ALL (81,82).
The blasts of ALL are typically negative for stains showing granulocytic differentiation, including Sudan black B, myeloperoxidase (MPO), and chloroacetate esterase. Precursor T-cell blasts may show cytoplasmic positivity for nonspecific esterase, partially inhibitable with sodium fluoride, usually found in a focal or dot pattern partially inhibited with sodium flouride. In addition, rare cases of ALL have been described showing weak positivity for Sudan black B and chloroacetate esterase. MPO may be demonstrable by immunophenotyping (see below).
Immunophenotyping
Immunophenotyping by flow cytometry is a key factor in the diagnosis and prognosis of ALL (83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116, 117, 118, 119, 120, 121, 122). Precursor B-cell immunophenotype is found in approximately 80% of both childhood and adult ALL, and precursor T-cell immunophenotype in most of the remainder. Immunophenotypic subtypes are further discussed below. In general, ALL shows aberrant antigen expression compared to normal precursor B-cells.
CD5 expression has been rarely reported in ALL and may confer a worse prognosis.
CD34 expression is found in approximately 50% of adult ALL and is associated with older age, hepatomegaly, myeloid antigen expression, and poor prognosis.
CD45 expression is found in approximately 90% of childhood ALL. CD45-negative cases are more likely to show precursor B-cell than the precursor T-cell phenotype.
CD56 expression, found in approximately 3% to 8% of ALL cases, is associated with FAB L2 morphology and central nervous system disease.
CD99 is expressed in ALL, but is also expressed in acute myeloid leukemia, Ewing sarcoma, and other tumors. In one reported case, the ALL was CD45-negative and CD99-positive, and was misdiagnosed as Ewing sarcoma. It should be kept in mind that ALL and Ewing sarcoma have been reported to coexist in some patients.
CD117 expression occurs in a minority of precursor B-cell and precursor T-cell ALL cases. The combined expression of CD13, CD33, and CD117 in ALL has been reported, and presents a problem in differential diagnosis.
Surface immunoglobulin light chain restriction may or may not be present. The expression of surface immunoglobulin is an indication of a more mature B-cell phenotype.
Terminal deoxynucleotidyl transferase (TdT) expression is found in more than 95% of ALL cases. Approximately 3% of ALL cases do not express TdT. These cases tend not to show CD10 or CD34 expression or MLL rearrangement. TdT-negative ALL may show either precursor B-cell or precursor T-cell immunophenotype.
Myeloid-associated antigen expression is found in approximately 30% to 35% of ALL cases and is directly correlated with precursor B-cell phenotype, t(9;22)(q34;q11), and rearrangements of MLL and ETV6. CD13 and CD33 are each expressed in approximately 15% of childhood ALL cases, CD15 in 7%, and CD14 in 1%. No prognostic significance is attached to the expression of myeloid antigens in ALL.
MPO expression and MPO mRNA may be found in precursor B-cell ALL, especially in cases with t(4;11)(4q21;q23) or t(9;22)(q34;q11). These findings are not necessarily accompanied by myeloid antigen expression.
Immunophenotype may be unstable in ALL. At presentation, a single clone may show multiple immunophenotypes. At relapse, approximately 50% of cases show immunophenotypic changes. Common changes include gain or loss of the myeloid antigens CD13 and CD33. Immunophenotype switching from lymphoid to myeloid immunophenotype and vice versa has been reported, especially in cases with t(4;11)(4q21;q23).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


