Myeloproliferative and Related Neoplasms
Diane C. Farhi
GENERAL FEATURES
Definition and Classification
Myeloproliferative neoplasms (MPN) are primarily characterized by increased peripheral blood cell counts, increased bone marrow cellularity, and preservation of hematopoietic maturation and differentiation (Table 21.1) (1, 2, 3, 4, 5, 6, 7). The 2008 World Health Organization classification is used in this discussion, with the addition of other syndromes that appear to be closely related (see Appendix A for WHO 2008 classification).
Classic MPN, recognized and classified in the 1950s, consists of polycythemia vera (PV), essential thrombocythemia (ET), primary myelofibrosis (PMF; formerly chronic idiopathic myelofibrosis and myelofibrosis with myeloid metaplasia), and chronic myelogenous leukemia (CML). The diagnosis was based largely on clinical findings, laboratory values, and bone marrow morphology.
With the discovery of the Philadelphia chromosome in 1960, CML was split off and eventually characterized by BCR’ABL1 translocation, thus separating the four classic entities into two groups: BCR’ABL1-positive disorders, namely CML; and BCR’ABL1-negative disorders, namely PMF, ET, and PV. The three BCR’ABL1-negative entities show many similarities and may be difficult to distinguish from one another. In addition, all four may show changes from one MPN phenotype or category to another within the same patient.
Other, less common, forms of MPN have been recognized over the years. These include chronic basophilic, eosinophilic, and neutrophilic leukemias; 8p11 myeloproliferative syndrome; mast cell disease (MCD); and possibly others. Mast cells are derived from hematopioetic stem cells and are therefore classified with MPN.
Molecular studies have played in a large role in the detection and classification of MPN. In many cases, a key gene is mutated or translocated, triggering constitutive kinase activity. This finding raises the possibility of targeted gene therapy not only in CML but also other MPN.
In general, the peripheral blood and bone marrow blast counts and myelodysplastic features are less marked in MPN than in myelodysplastic syndromes (MDS); however, no clear criteria divide all MPN and MDS cases. Cases with features of both are discussed as MDS’MPN (see Chapter 20).
Clinical Course
MPNs usually present with the insidious onset of non-specific complaints, abdominal pain referable to hepatosplenomegaly and’or coagulopathy. Many patients are initially asymptomatic and the diagnosis is made after routine peripheral blood screening. The course is generally indolent in the initial chronic phase of disease, followed by a period of decreasing peripheral blood cell counts and increasing blast percentage (accelerated phase), with eventual transformation to myelodysplastic syndrome (MDS) or acute leukemia (blast crisis). Progression to acute leukemia occurs with a variable frequency, depending on the specific type of MPN.
Many types of MPN have been reported with lymphoid, plasma cell, and histiocytic neoplasms, both clonally related and unrelated. The clonally related neoplasms occur as a consequence of lymphoid, plasma cell, or histiocytic differentiation of the neoplastic clone.
Peripheral Blood and Bone Marrow Findings
The peripheral blood counts are variable but usually include at least one cell line that is increased. Neutrophilia, eosinophilia, basophilia, and thrombocytosis are common. Platelets often appear enlarged, abnormally shaped, and’or hypogranular. The blast percentage in the peripheral blood and bone marrow is less than 3% in the chronic phase, higher in the accelerated phase, and 20% or more in leukemic transformation.
The bone marrow is typically hypercellular and shows hematopoietic maturation. Megakaryocytic hyperplasia and dysmegakaryopoiesis are characteristic. Hypervascularity, reticulin fibrosis, and osteosclerosis are frequently found. For unknown reasons, stainable iron stores are often absent; this finding does not indicate iron deficiency. Bone marrow findings tend to change over time, with increasing dysmyelopoiesis, more blasts, and worsening fibrosis and osteosclerosis.
Immunophenotyping
Immunophenotyping is of limited value until an accelerated phase or conversion to acute leukemia occurs, when blast enumeration and immunophenotyping may be helpful. In
the chronic phase, the peripheral blood shows a nonspecific increase in CD34-positive stem cells.
the chronic phase, the peripheral blood shows a nonspecific increase in CD34-positive stem cells.
TABLE 21.1 WHO 2008 Classification of Myeloproliferative Neoplasms | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
Genetic Analysis
Genetic analysis by karyotyping and molecular methods have yielded important information in MPN (8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23). The most significant genes are ABL1, BCR, FIP1L1, PDGFRA, PDGFRB, JAK2, KIT, and other genes either encoding protein kinases or involved as translocation partners with protein kinase genes. Anomalies of kinase genes leads to constitutive kinase activation, a key process in MPN.
Clonality can be shown, by definition, in all cases of chronic myelogenous leukemia (CML) by demonstration of BCR’ABL1 or a variant, and in approximately 50% of cases of PMF, ET, and PV by JAK2 mutation. Although clonal anomalies may be present at diagnosis or appear with time, they appear to be the result of clonal evolution and not a specific pathogenetic event. At present, evidence indicates that many cases show polyclonal hematopoiesis, with or without a concurrent clonal anomaly. With duration of disease, BCR’ABL1-negative neoplasms progress from nonclonal, to both nonclonal and clonal, and finally to clonal hematopoiesis.
BCR’ABL1 fusion and variants have been extensively studied. Any one of at least three different breakpoints in BCR are known. These yield different fusion proteins and different clinical syndromes. Different BCR breakpoints and fusion proteins are found in (a) CML, (b) an atypical form of CML with monocytic differentiation, and (c) chronic neutrophilic leukemia. BCR rearrangement is not limited to these disorders but has been shown in other hematopoietic disorders.
Evidence suggests that BCR’ABL1 is a sign of clonal evolution rather than an initial pathogenetic event, as BCR’ABL1-negative clonal populations have been identified in cases of CML. Furthermore, the presence of BCR’ABL1 alone is not sufficient to cause CML, as this translocation has been reported in hematopoietic cells of individuals without clinical evidence of disease. PMF, ET, and PV do not show BCR’ABL1 translocation, except when rarely acquired in the course of clonal evolution.
Mutation of JAK2, located on chromosome 9p24, is present in nearly 100% of PV cases, and in approximately 50% of ET and PMF cases, specifically the V617F mutation. In a minority of cases, a JAK2 mutation at exon 12, JAK2 rearrangement, or an MPL mutation has been identified. CD177 (previously PRV1) is overexpressed but not mutated in nearly all cases of ET and PV. JAK2 mutation is also associated with endogenous erythroid colony formation by cultured hematopoietic cells.
It should be kept in mind that JAK2 mutation is not restricted to PMF, ET, and PV, but has also been demonstrated in mast cell disease, chronic myelomonocytic leukemia, hypereosinophilic syndrome, chronic neutrophilic leukemia, and B-cell lymphoma.
Extramedullary Disease
MPN is frequently, if not always, accompanied by extramedullary disease. The liver, spleen, and lymph nodes are most often affected but virtually any organ or body cavity may be involved. Extramedullary disease may occur at any stage of MPN. The histologic findings in the extramedullary site may reflect the chronic phase of disease, accelerated phase, or leukemic transformation, regardless of concurrent bone marrow histology.
Differential Diagnosis
MPN in the chronic phase may be difficult to distinguish from reactive hematopoietic proliferations. MPN in the accelerated phase may resemble myelodysplastic syndromes. MPN with leukemic transformation should be distinguished from de novo acute leukemia, therapy-related acute leukemia, and malignant lymphoma.
PRIMARY MYELOFIBROSIS
Primary myelofibrosis (PMF; previously known as chronic idiopathic myelofibrosis and myelofibrosis with myeloid metaplasia), is characterized by anemia and leukoerythroblastosis (Figs. 21.1, 21.2, 21.3and 21.4) (24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36). It occurs predominantly in middle-aged to older adults. PMF has been diagnosed in children but it is possible that in these rare cases, the disorder represents a congenital bone marrow failure syndrome, acute leukemia with fibrosis, or other disorder.
PMF occurs as a de novo disorder and as a late stage of PV and ET.
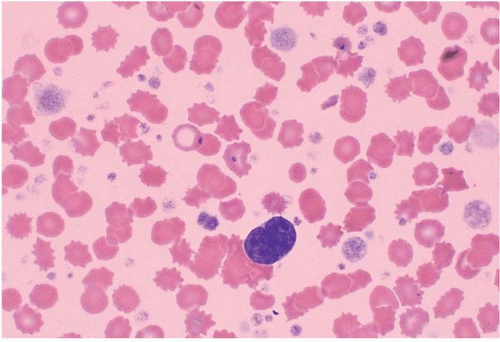 FIG. 21.1 Primary myelofibrosis, peripheral blood. A micromegakaryocyte is present, surrounded by increased and abnormal platelets. |
Clinical signs and symptoms are referable to cytopenias, coagulopathy, and excessive extramedullary hematopoiesis. Palpable splenomegaly is found in approximately 75% and hepatomegaly in almost 50% of patients at diagnosis. Massive hepatosplenomegaly occurs late in the disease. The clinical course is usually progressive, with worsening cytopenias and eventual transformation to myelodyplastic syndrome (MDS) or acute leukemia. Rarely, PMF converts to PV. Survival is significantly worse than in PV or ET. The risk of transformation to acute leukemia is related to the type and duration of therapy and is increased by splenectomy. Spontaneous hematologic remission rarely occurs, followed in most cases by leukemic transformation. Survival up to 20 years after diagnosis has been reported.
Laboratory studies usually show normocytic normochromic anemia with an increased red cell distribution width (RDW). The white blood cell (WBC) and platelet counts are variable, and may be either increased or decreased. The majority of cases show neutrophilia and basophilia at diagnosis.
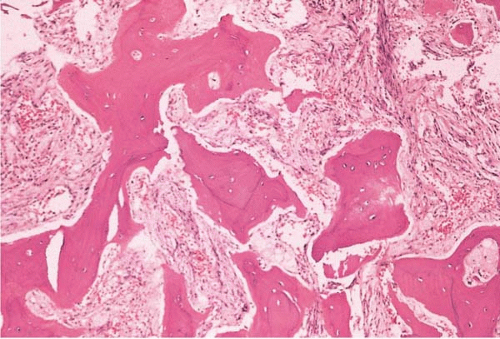 FIG. 21.2 Primary myelofibrosis, bone marrow biopsy. Osteosclerosis and marrow fibrosis are prominent. |
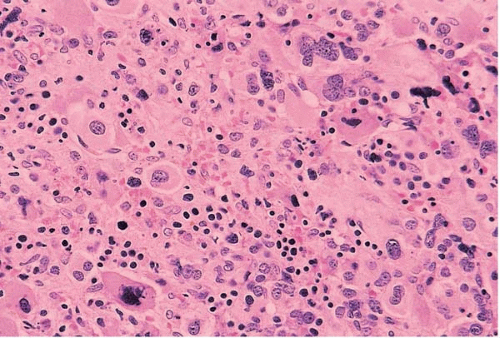 FIG. 21.3 Primary myelofibrosis, bone marrow biopsy. Cellularity is virtually 100% in the early, or cellular, phase of the disease. Abnormal megakaryocytes are prominent. |
Peripheral blood smears characteristically show teardrop red blood cells and significant aniso- and poikilocytosis. Other findings include immature neutrophils and abnormal platelets, particularly large, irregular, hypogranular platelets. Naked megakaryocytic nuclei and fragments of megakaryocytic cytoplasm are often present. Granulocytes and precursors may show mild dysplastic changes. Basophilia and eosinophilia are often present. Over time, neutropenia, thrombocytopenia, and circulating blasts appear.
Bone marrow aspirate smears may be hypercellular in the early stages of PM and hypocellular in patients with advanced fibrosis. Most patients are diagnosed at the fibrotic stage. Megakaryocytes are increased and atypical, varying in size, maturation, nuclear shape, and nuclear lobation. Stainable iron may be absent, a finding that does not necessarily indicate iron deficiency.
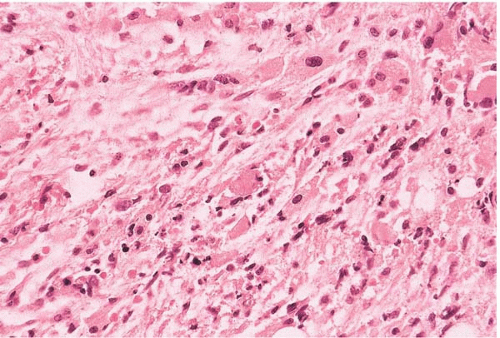 FIG. 21.4 Primary myelofibrosis, bone marrow biopsy. Fibrous tissue compresses atypical megakaryocytes and other residual hematopoietic cells. This appearance is indistinguishable from the terminal phase of polycythemia vera or essential thrombocythemia. |
Histologic sections show variable cellularity, depending on the stage of disease. Most cases are advanced at diagnosis and show hematopoietic hypocellularity. The myeloid-to-erythroid (M:E) ratio and blast percentage are often difficult to determine because of compression of hematopoietic cells by marked fibrosis. Megakaryocytes are increased, distributed singly and in clusters, and atypical, with varying size and shape, bizarre forms, enlarged and hyperchromatic nuclei, and abnormal nuclear lobation.
Stromal changes are usually marked. Reticulin fibrosis is present because of local cytokine secretion and proliferation of polyclonal fibroblasts. In advanced cases, collagen fibrosis may be found. Changes in trabecular bone parallel the degree of fibrosis. The trabeculae are thickened and irregular, sometimes to the point of overt osteosclerosis. Other findings include benign lymphoid aggregates, sea-blue histiocytosis, gelatinous transformation, and granulomas. Therapy may reduce the degree of fibrosis.
Early or cellular-stage PMF may be difficult to recognize and classify. Patients present with minimal to modest splenomegaly. The peripheral blood shows mild leukocytosis, therapy-refractory anemia with occasional teardrop forms, thrombocytosis, and relatively few myeloid and erythroid precursors. The bone marrow is hypercellular with an increased myeloid to erythroid (M:E) ratio, increased immature myeloid cells, increased and atypical megakaryocytes, and little or no fibrosis. Cases with these features may be diagnosed as unclassifiable MPD until findings more characteristic of PMF emerge.
Karyotype studies show clonal karyotypic anomalies in approximately 50% of patients, primarily trisomies of chromosome 1q, 8, and 9, and deletions of chromosomes 12p, 13q14, and 20q. Acquisition of additional anomalies is evidence of clonal progression and presages leukemic transformation. BCR’ABL1 translocation is typically absent but has been rarely reported in the course of clonal evolution. It is possible that some PMF cases with BCR’ABL1 are actually examples of CML with marked fibrosis.
Molecular studies show JAK2 V617F mutation in approximately 40% to 50% of patients. JAK2, located on chromosome 9p24, encodes a tyrosine kinase, and this mutation produces constitutive kinase function. Cases with JAK2 mutation tend to show higher WBC and red blood cell counts, lower platelet counts. Such cases also tend to show less prominent megakaryocytic clustering. Cases lacking JAK2 V617F mutations may show mutation of JAK2 exon 12 or MPL, located on chromosome 1p34, which encodes the thrombopoietin receptor. PMF may show either mutated or nonmutated PM; thus, the mutations found to date do not seem to be the initial neoplastic event in PMF.
PMF has been reported with sarcoidosis, amyloidosis, and clonal B-cell and plasma cell neoplasms.
The differential diagnosis includes reactive marrow fibrosis, other MPN, MDS, acute leukemia with fibrosis, and lymphoid and plasma cell neoplasia with fibrosis. In general, PMF is distinguished from PV by a lower hematocrit, more atypical megakaryocyte morphology, and more prominent fibrosis; from ET by more atypical megakaryocyte morphology and prominent fibrosis; and from CML by the absence of BCR’ABL1 translocation. It may be difficult to distinguish PMF from MDS with fibrosis. In general, PMF shows a more insidious clinical course, more prominent splenomegaly, less impressive dysplasia, and fewer cytogenetic anomalies.
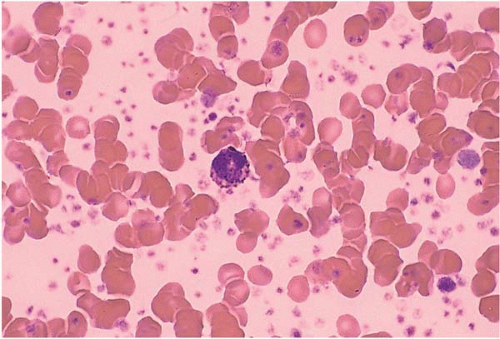 FIG. 21.5 Essential thrombocythemia, peripheral blood. A basophil is surrounded by numerous platelets, some of which show abnormal shapes and hypogranularity. |
ESSENTIAL THROMBOCYTHEMIA
Essential thrombocythemia (ET) is characterized primarily by thrombocytosis (Figs. 21.5, 21.6, 21.7and 21.8) (37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64). ET occurs as a de novo disorder, a therapy-related disease, and a transformation of polycythemia vera (PV).
ET occurs in both children and adults, more often in females. Clinical signs and symptoms are referable to coagulopathy
and excessive hematopoiesis, and include thromboembolism and mild hepatosplenomegaly. Many patients are asymptomatic. The clinical course is usually indolent, with increased fibrosis and osteosclerosis; acute leukemia occurs in approximately 1% to 5% of patients. The risk of transformation is related to the type and duration of therapy. Spontaneous remission has been reported during pregnancy.
and excessive hematopoiesis, and include thromboembolism and mild hepatosplenomegaly. Many patients are asymptomatic. The clinical course is usually indolent, with increased fibrosis and osteosclerosis; acute leukemia occurs in approximately 1% to 5% of patients. The risk of transformation is related to the type and duration of therapy. Spontaneous remission has been reported during pregnancy.
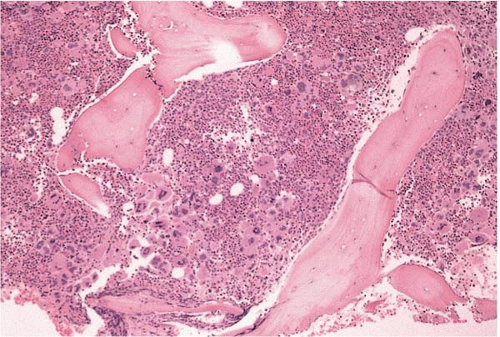 FIG. 21.6 Essential thrombocythemia, bone marrow biopsy. Osteosclerotic bone surrounds hypercellular bone marrow with increased and multilobated megakaryocytes. |
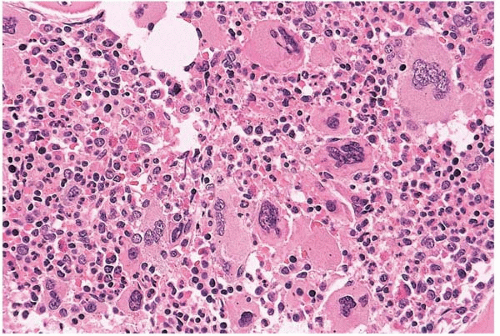 FIG. 21.7 Essential thrombocythemia, bone marrow biopsy. Megakaryocytes are increased, multilobated, and atypical; the intervening cells are a mixture of red and white blood cell precursors. |
Laboratory studies reveal thrombocytosis, neutrophilia, and normal to decreased hematocrit. The neutrophil alkaline phosphatase score is increased. Mild eosinophilia and basophilia are usually present. The platelet count is usually more than 500 × 109‘L, but symptomatic ET has been reported with platelet counts as low as 300 × 109‘L. ET usually shows of 500-2000 × 109‘L. Platelet function studies are often abnormal. Serum thrombopoietin is normal to increased. Circulating hematopoietic precursors capable of spontaneous erythroid colony formation are increased; however, in practice, this is a difficult test to perform reliably and is neither sensitive nor specific enough to use as a diagnostic criterion.
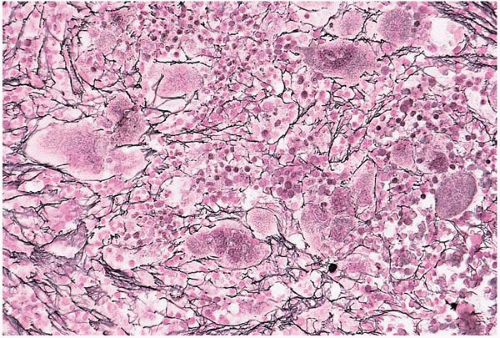 FIG. 21.8 Essential thrombocythemia, bone marrow biopsy. Reticulin fibers are increased and outline many of the abnormal megakaryocytes (silver stain for reticulin). |
The peripheral blood smear shows normal red blood cell morphology in uncomplicated ET. Neutrophils may be morphologically normal or slightly dysplastic, with subtle abnormalities and increased nuclear lobation. Platelets are often normal in appearance but may include large and hypogranular or agranular forms.
Bone marrow aspirate smears show a variable M:E ratio. Hematopoietic maturation is essentially unremarkable in the untreated patient. Blasts typically comprise less than 3% of nucleated cells. Megakaryocytes are plentiful and mature and include large forms with hyperlobated nuclei. Iron stores are decreased to absent, a finding that does not necessarily reflect true iron deficiency.
Histologic sections of the bone marrow show increased cellularity. This is prominent in megakaryocytes, which are usually present in clusters. Megakaryocytes range from abnormally small to very large, with larger cells showing increased atypical nuclear contours, abnormal nuclear lobation, and densely clumped or almost pyknotic chromatin. Reticulin fibrosis may be present. The trabeculae show irregular thickening and thinning. Other reported findings include pure red cell aplasia, necrosis (possibly because of antiphospholipid antibody syndrome), and reactive histiocytosis.
Genetic studies show monoclonality in some, but not all, cases. Cases with monoclonality (as evidenced by chromosome X inactivation or other means) tend to show overexpression of CD177 (formerly PRV1); the significance of this finding is not known. The appearance of clonal karyotypic anomalies indicates clonal evolution, and correlates with an increased risk of leukemic transformation. Commonly acquired anomalies include deletion of chromosome 17p and’or P53 mutation. BCR’ABL1 translocation is absent. The JAK2 V617F gain-of-function mutation is found in approximately 50% of patients. Such cases tend to show higher white blood cell and red blood cell counts, as well as lower platelet counts and less prominent megakaryocytic clustering.
ET may transform to PV, PMF, and systemic mastocytosis.
ET has been reported with sarcoidosis and with clonally related and unrelated B-cell and plasma cell neoplasms.
The differential diagnosis includes reactive thrombocytosis; other MPN, especially PV, the early (cellular) stage of PMF, and CML with thrombocytosis; and MDS with thrombocytosis. ET is distinguished from PV primarily by a higher platelet count and lower hematocrit; from PMF by thrombocytosis and less marked fibrosis; and from CML by the absence of BCR’ABL1 translocation. It may not always be possible to distinguish ET from PV or from MDS with thrombocytosis.
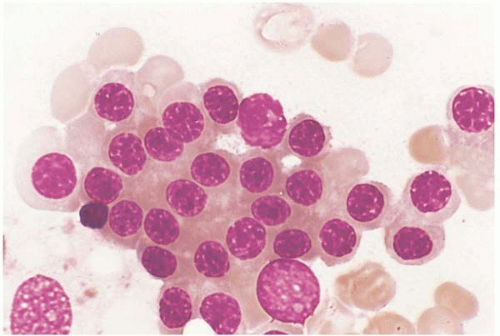 FIG. 21.9 Polycythemia vera, bone marrow aspirate. Numerous erythroid precursors are present, showing no discernible morphologic abnormalities. |
POLYCYTHEMIA VERA
Polycythemia vera (PV) is characterized primarily by erythrocytosis (Figs. 21.9, 21.10, 21.11, 21.12, 21.13and 21.14) (65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94). PV occurs as a de novo disorder, a therapy-related disease, and a transformation of (or in conjunction with) primary myelofibrosis (PMF), essential thrombocythemia (ET), systemic mastocytosis, and chronic myelogenous leukemia (CML).
PV usually occurs in middle-aged and older adults, rarely in adolescents, and more often in males. Clinical signs and symptoms are referable to hyperviscosity, coagulopathy, and excessive hematopoiesis. These include arterial and venous thromboembolism, vasculopathy, neurologic deficits, and mild hepatosplenomegaly. Many patients are asymptomatic.
The clinical course is usually indolent, with eventual transformation to fibrosis and osteosclerosis, another MPN, MDS, or acute leukemia in approximately 1% to 5% of patients. The risk of transformation is related to the type and duration of therapy. Spontaneous remission has been rarely reported.
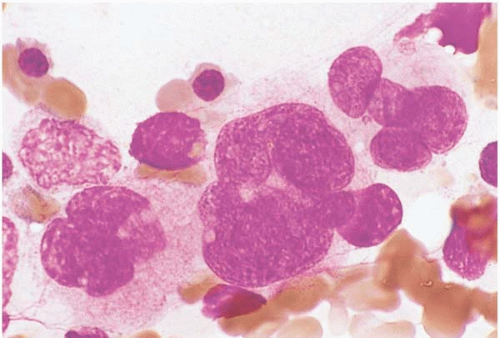 FIG. 21.10 Polycythemia vera, bone marrow aspirate. Megakaryocytes are increased and show numerous nuclear lobations. |
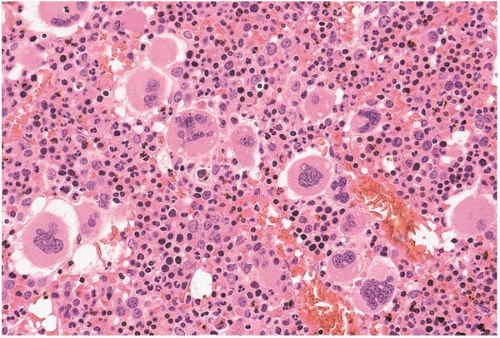 FIG. 21.11 Polycythemia vera, bone marrow biopsy. Megakaryocytes are increased and multilobated; erythroid precursors are also increased. |
Laboratory studies reveal an increased packed red blood cell volume or hematocrit (>60% in males, >56% in females), neutrophilia (>10 × 109‘L in nonsmokers), and thrombocytosis (>400 × 109‘L). The neutrophil alkaline phosphatase score is increased. Mild eosinophilia and basophilia are usually present. The red blood cell mass is increased but is difficult to measure and may be falsely low due to obesity. Serum erythropoietin is decreased. Circulating hematopoietic precursors capable of endogenous erythroid colony formation are increased. However, in practice this is a difficult test to perform reliably, and it is neither sensitive nor specific enough to use as a diagnostic criterion.
Red blood cell and platelet counts may be affected in PV by concomitant hematopoietic disorders or other factors. The platelet count may be increased by iron deficiency and the hematocrit increased by hypoxia or renal disorders. Conversely, the hematocrit may be decreased by expanded
plasma volume, megaloblastic anemia, iron deficiency, or renal failure.
plasma volume, megaloblastic anemia, iron deficiency, or renal failure.
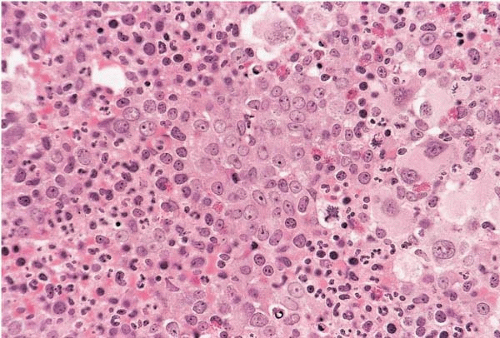 FIG. 21.12 Polycythemia vera with focal transformation to acute myeloid leukemia, bone marrow biopsy. An interstitial collection of blasts is present. |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


