Hemoglobin and myoglobin are chemically similar molecules that contain porphyrin groups. Porphyrins are cyclic structures comprised of four pyrrole rings joined by methine bridges. The arrangement of the pyrrole nitrogen atoms in the porphyrin ring allows chelation of a metal ion to form functional groups that participate in oxidative metabolism. The chelation of ferrous iron to protoporphyrin produces heme, the prosthetic group of hemoglobin responsible for binding oxygen. The analysis of porphyrins in the laboratory aids in the diagnosis of a group of rare disorders called porphyrias that result from disturbances in heme synthesis.
Hemoglobin molecules are designed to bind, deliver, and release oxygen. Qualitative defects in the hemoglobin molecule result in a group of disorders called hemoglobinopathies. Quantitative defects in the production of hemoglobin molecules lead to another group of disorders, called thalassemias. Myoglobin is a simple heme protein found in cardiac and skeletal muscles. Damage to the heart or muscle releases myoglobin into the blood. Increased serum myoglobin is seen following acute myocardial infarction (AMI), muscle injury, and intense exercise.
PORPHYRINS
Porphyrins are chemical intermediates in the synthesis of hemoglobin, myoglobin, and other respiratory pigments called cytochromes. Iron is chelated to the porphyrin ring to form heme, which is incorporated into proteins to produce biologically functional hemoproteins, such as hemoglobin, myoglobin, cytochrome P450, and the peroxidase and catalase enzymes.1 Porphyrins are analyzed in clinical chemistry to aid in the diagnosis of porphyrias, diseases that result from disturbances in heme synthesis. Excess amounts of these intermediate compounds in blood, urine, or feces indicate a metabolic block in heme synthesis.
Porphyrin Properties
There are many naturally occurring porphyrins. The pigment chlorophyll is a magnesium porphyrin that enables plants to use light energy to synthesize carbohydrates. Most porphyrins in the human body exist as porphyrinogens, reduced compounds that are intermediates in the biosynthesis of heme, the nonprotein, iron-containing prosthetic group of hemoglobin. Porphyrinogens are oxidized on exposure to air to porphyrins. Substitution of the pyrrole rings that make up the porphyrinogen macrocycle generates four isomers for every porphyrin compound; however, only type I and type III isomers occur naturally. These isomers differ in the spatial arrangement of the pyrrole substituents.2 Only type III isomers form heme; however, in some disorders, the nonfunctional type I isomers may be found in excess in body fluids and tissue.
Porphyrins are photoactive due to extensive conjugation of the tetrapyrrole ring. Their dark red color is due to a strong absorbance in the visible region of the spectrum. The conjugated tetrapyrrole ring is highlighted in the diagram of protoporphyrin shown in Figure 19.1. The compounds absorb light of 400-nm wavelength and emit a characteristic orange-red fluorescence between 600 and 650 nm.3 This property produces some of the manifestations of disease and provides a means for detection of the compounds in body fluids tested in the laboratory. Aqueous solubility of porphyrins varies with the number of carboxylic acid substituents present in the particular porphyrin compound. Uroporphyrin (Uro) has eight carboxylic acid groups, is the most soluble in water, and is excreted renally. Porphyrins with only two carboxyl groups are the least soluble. These compounds are present in blood and excreted in the feces. Coproporphyrin (Copro) has four carboxylic acid substituents and intermediate solubility. Coproporphyrins are found in blood, urine, and feces. Solubility influences the type of specimen selected for the measurement of particular porphyrin intermediates.
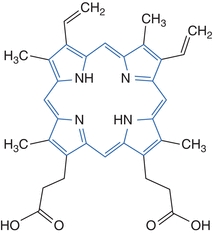
Figure 19.1 Structure of porphyrins: protoporphyrin.
CASE STUDY 19.1
A 58-year-old man with a history of alcoholism complained of increased skin fragility and skin lesion formation on his hands, forehead, neck, and ears on exposure to the sun. On physical examination, hyperpigmentation and hypertrichosis were observed. Laboratory testing of a 24-hour urine specimen showed increased uroporphyrin, heptacarboxyl porphyrin, and a slight increase in coproporphyrin; testing for ALA and PBG was negative. Isocoproporphyrin was elevated in the feces. Serum ferritin and serum transaminase were increased.
questions
1. What is the most probable disorder?
2. Which confirmatory test should be ordered?
3. What is the most probable precipitating factor?
4. What are other causes of acquired cases of this type of porphyria?
5. How is this type of porphyria differentiated from other porphyrias causing cutaneous symptoms?
CASE STUDY 19.2
A few days after a laparotomy for “intestinal obstruction,” a young nurse from South Africa became emotionally disturbed and appeared to be hysterical. For more than 1 week before the surgery, she had taken barbiturate capsules as a sleep aid. She complained of severe abdominal and muscle pain and general weakness. Her tendon reflexes were absent, and she was vomiting and constipated. Her urine was dark in color on standing and gave a brilliant pink fluorescence when viewed in ultraviolet light. Within 24 hours, she was completely paralyzed, and after 2 days she expired.
questions
1. What possible condition did this young woman have, and why did it manifest at this time?
2. Would members of her family be at risk for a similar disease?
3. Which enzyme defect should be suspected?
4. What other confirmatory tests, if any, could be performed?
Biochemistry: Synthesis of Heme
Although all cells contain hemoproteins and can synthesize heme, the bone marrow and liver are the primary sites for heme production.2 Biosynthesis is accomplished in a sequence of reactions catalyzed by eight different enzymes in which one molecule of heme is produced from eight molecules of δ-aminolevulinic acid (ALA). The first and last three synthetic reactions require a significant amount of energy and take place in the mitochondrion. Other reactions, which do not require as much energy, occur in the cytosol. Each reaction in the pathway is catalyzed by a particular enzyme. Heme biosynthesis is diagrammed in Figure 19.2.
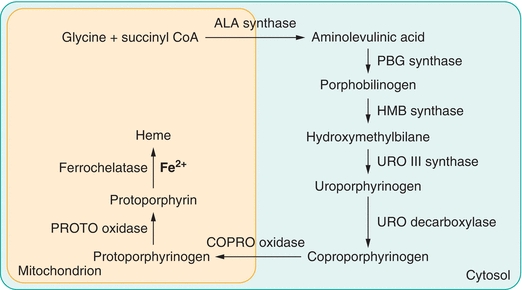
Figure 19.2 Heme biosynthesis.
In the first step in the pathway, glycine and succinyl coenzyme A are condensed to form δ-aminolevulinic acid. In the next reaction, the enzyme porphobilinogen synthase catalyzes condensation of two molecules of ALA to form the monopyrrole porphobilinogen (PBG). Polymerization of four molecules of PBG creates a linear tetrapyrrole, hydroxymethylbilane. This process is catalyzed by the enzyme hydroxymethylbilane synthase (also called PBG deaminase). The enzyme uroporphyrinogen III synthase catalyzes an intramolecular rearrangement and ring closure. The product of this reaction is the cyclic isomer, uroporphyrinogen III. Sequential removal of one carboxyl group from each pyrrole ring in the cyclic tetrapyrrole is catalyzed by uroporphyrinogen decarboxylase. The hepta-, hexa-, penta-, and tetra-carboxyl intermediates are formed in succession. In subsequent reactions, coproporphyrinogen oxidase catalyzes the oxidation of two additional carboxylic acid substituents to form protoporphyrinogen IX, which is oxidized to protoporphyrin (Proto) in a reaction catalyzed by protoporphyrinogen oxidase. In the final step of biosynthesis, ferrochelatase catalyzes the insertion of ferrous iron into the porphyrin ring to form heme.
Heme formation is regulated by negative feedback. Control of the rate of heme synthesis in cells in the liver is achieved largely through regulation of the enzyme δ-aminolevulinic acid synthase (ALAS). Heme inhibits synthesis of the constitutive gene ALAS1 at transcription and translation and also controls transfer from the cytosol to mitochondria.4 Increases in the pool of hepatic heme diminish the production of ALAS (a negative feedback mechanism). Conversely, ALAS production is increased with a depletion of heme. The size of the regulatory heme pool is influenced by the requirement for hemoproteins in the liver. Drugs and other compounds appear to induce ALAS synthesis by several different mechanisms, but all result in depletion of the regulatory heme pool. Therefore, the rate of heme synthesis can change rapidly in response to a variety of external stimuli. In bone marrow erythrocytes, other enzymes in the pathway and the rate of cellular iron uptake seem to control the rate of heme synthesis.5
Spontaneous Formation of Porphyrin Isomers
When the intracellular concentration of PBG is sufficiently high, polymerization occurs without enzymatic catalysis. If the enzyme hydroxymethylbilane synthase is deficient, PBG accumulates and PBG monomers polymerize to the linear tetrapyrrole, hydroxymethylbilane, which spontaneously cyclizes. There is no intramolecular rearrangement and a different isomer, uroporphyrinogen I is formed.2,4 Uroporphyrinogen I is a substrate for uroporphyrinogen decarboxylase and four carboxyl groups can be removed to produce coproporphyrinogen isomer I. Coproporphyrinogen oxidase cannot oxidize coproporphyrinogen I, which accumulates and is excreted. Deficiency of uroporphyrinogen III synthase allows hydroxymethylbilane to accumulate and cyclize to uroporphyrinogen I, which is decarboxylated to Copro I. In this case, too, coproporphyrinogen I accumulates and is excreted. The porphyrin isomers Uro I and Copro I are found in biological fluids from patients with particular porphyrias.
Pathophysiology: Disorders of Heme Biosynthesis
The porphyrias are a group of rare inherited or acquired metabolic disorders caused by loss or gain of function mutations in the enzymes responsible for heme biosynthesis.6 The defective enzyme results in overproduction, accumulation, and excretion of toxic precursor compounds and porphyrins. Conditions corresponding to enzyme abnormalities have been identified in every step of heme synthesis. Most of the diseases have an autosomal dominant inheritance pattern (mutation of one gene causes disease), and there is a 50% reduction in activity of the affected enzyme. Some patients demonstrate an enzyme deficiency but do not experience clinical or biochemical manifestations of porphyria, indicating that other factors, such as demand for increased heme biosynthesis, operate to cause disease expression.
Deficient activity of the first enzyme in the pathway, ALA synthase, is associated with an X-linked recessive disorder and sideroblastic anemia—not a porphyria. An X-linked dominant gain-of-function mutation in the erythroid-specific ALAS enzyme results in increased enzyme activity and production of excess protoporphyrin.7 Deficient activities of any of the other enzymes in the pathway cause a porphyria characterized by excessive accumulation and excretion of intermediate compounds that produce particular symptoms. In most cases, residual enzyme activity produces sufficient heme to prevent anemia. Autosomal recessive diseases also occur; these conditions are associated with more severe symptoms. Late-onset porphyria has been reported as a rare complication of hematologic disorders.8,9
Porphyrias can be grouped according to the clinical manifestations of disease.6,10 An excess of the early precursors in the pathway of heme synthesis (ALA, PBG) causes neuropsychiatric symptoms, including abdominal pain, nausea, constipation, hypertension, psychiatric symptoms, fever, and paresthesia. Porphyrias in this category include ALA dehydratase deficiency porphyria (ADP) and acute intermittent porphyria (AIP). Excesses of porphyrin intermediates produce cutaneous symptoms of photosensitivity, blisters, excess facial hair, and hyperpigmentation. Congenital erythropoietic porphyria (CEP), porphyria cutanea tarda (PCT), erythropoietic protoporphyria (EPP), and X-linked protoporphyria (XLPP) are associated with cutaneous symptoms. Porphyrin-induced photosensitivity manifests through increased fragility of light-exposed skin, in CEP and PCT, or by burning of light-exposed skin, in EPP and XLPP. The photosensitizing effects of the porphyrins are attributed to absorption of light. Excesses of both precursor compounds and porphyrin intermediates cause neurocutaneous symptoms that are found in hereditary coproporphyria (HCP) and variegate porphyria (VP).
Conditions associated with neurological symptoms and serious acute attacks are termed acute porphyrias. The acute porphyrias are ADP, AIP, HCP, and VP. AIP is the most common acute porphyria, although most (~80%) individuals with a genetic defect are asymptomatic. Attacks usually begin in early adulthood, occur more often in women, and may be precipitated by a variety of factors. Severe neurological attacks can be life threatening. In HCP and VP, ineffective catalysis by coproporphyrinogen oxidase or protoporphyrinogen oxidase results in inhibition of hydroxymethylbilane synthase and consequent increase in PBG concentrations. For this reason, HCP and VP are classified as acute porphyrias. In these diseases, porphyrin intermediates accumulate also and symptoms associated with photosensitivity may occur.
The cutaneous (nonacute) porphyrias present as chronic conditions. Exposure to sunlight results in photosensitization of the skin due to accumulation of excess porphyrins in tissues. The porphyrins absorb light and react, producing blistering skin lesions on sun-exposed areas that characterize the disease. CEP, PCT, EPP, and XLPP are cutaneous porphyrias. PCT is the most common cutaneous porphyria and occurs more often as a sporadic condition. Exogenous factors including alcohol consumption, estrogen use, hepatitis C or HIV infection, and increased hepatic iron content, as occurs in hemochromatosis, may precipitate development of PCT. The autosomal recessive disease, CEP, is associated with extreme photosensitivity. Exposure to sunlight destroys the skin and may cause disfiguring changes to facial features, fingers, and toes, and corneal scarring. Hepatoerythropoietic porphyria (HEP) is a homozygous form of PCT characterized by severe clinical symptoms, which are similar to CEP. In EPP, deficiency of ferrochelatase causes accumulation of protoporphyrin in red blood cells, plasma, and feces. Redness, edema, and painful burning sensations occur when skin is exposed to sunlight. Disease onset happens early in life, and porphyria may be overlooked as the cause of these symptoms in a young child. XLPP manifests as increased activity of the erythroid-specific ALAS enzyme and massive production of protoporphyrin with consequent cutaneous symptoms similar to those found in EPP.
Treatment of porphyria is directed at modifying the biochemical abnormalities causing clinical symptoms. Intravenous hematin is used to counteract acute attacks of neurologic dysfunction. Hematin inhibits the enzyme ALAS1 and reduces porphyrin synthesis. Drugs and medications known to precipitate acute attacks must not be used. Cutaneous symptoms are managed by minimizing sun exposure, use of protective clothing and topical sun-blocking agents, and oral administration of β-carotene, which acts as a singlet oxygen trap, to prevent skin damage. Application of the synthetic α-melanocyte-stimulating hormone analogue, afamelanotide, provides photoprotection by increasing melanin in the skin.11 Phlebotomy, to reduce iron stores, and low-dose chloroquine, to increase porphyrin excretion, are used to treat PCT. Precipitating factors, such as ingestion of alcohol or estrogens, should be avoided. Blood transfusion may be necessary in CEP and HEP. Severe cases of porphyria have been treated effectively with bone marrow or liver transplantation.6
Secondary Porphyrinurias
Secondary porphyrinurias are acquired conditions associated with increased excretion of urinary porphyrins.3 The disorders are not the result of an inherited biochemical defect in heme synthesis but are due to another disorder or toxin interfering with heme formation or metabolism. Liver disease and heavy metal poisoning can cause excess urinary porphyrin excretion in the absence of porphyria. Impaired heme synthesis may result from functional changes due to liver diseases such as hepatitis, cirrhosis, and obstructive jaundice.12 The toxic metal lead inhibits enzymatic formation of PBG, increasing concentrations of ALA, and coproporphyrinogen oxidase function, which causes Copro III to accumulate. Lead interferes with insertion of ferrous iron into heme, and an alternate product, zinc protoporphyrin, is formed by incorporation of zinc ions into the protoporphyrin ring. Some symptoms of lead intoxication are similar to those found in acute porphyria.
Biliary excretion of coproporphyrin isomers is altered in the inherited disorders of bilirubin metabolism (Dubin Johnson, Rotor, and Gilbert syndromes).13 In Dubin Johnson syndrome, excretion of Copro I is increased and Copro III excretion is decreased. Total urinary coproporphyrin may be slightly elevated, and the ratio of Copro I to Copro III is greater than 80%. In Rotor syndrome, there is a shift from biliary to urinary porphyrin excretion, and total urinary Copro is increased. Copro I accounts for about 70% of the total amount. In Gilbert’s syndrome, 50% of urinary coproporphyrin is the Copro I isomer. These ratios are the opposite of the expected coproporphyrin ratio. In unaffected individuals, more Copro III than Copro I is excreted in the urine; the Copro I: Copro III isomer ratio is approximately 30%:70%.14,15
Clinical Application
Testing for porphyrins and porphyrin precursors is used to diagnose and monitor porphyrias and other disorders of porphyrin metabolism. Methods available for identification of porphyrias include genetic assays to detect DNA mutations, functional and quantitative assays for the enzymes responsible for heme biosynthesis, and biochemical assays to identify and quantify porphyrins and precursor compounds. Only a few specialized laboratories perform molecular genetic testing and enzyme assays.16 Clinical diagnostic tests for porphyrin disorders are based most often on identification of characteristic patterns of excess porphyrin precursor and intermediate compounds in body fluids (Table 19.1). The specimen tested (urine, blood, or feces) depends on solubility and excretion of the metabolic intermediates associated with particular disease symptoms. Acute symptoms indicate testing for the porphyrin precursors PBG and ALA in urine. Patients with cutaneous photosensitivity are evaluated by examination of blood, urine, and feces for excess porphyrin intermediates.
TABLE 19.1 Heme Biosynthesis: Metabolites and Diseases
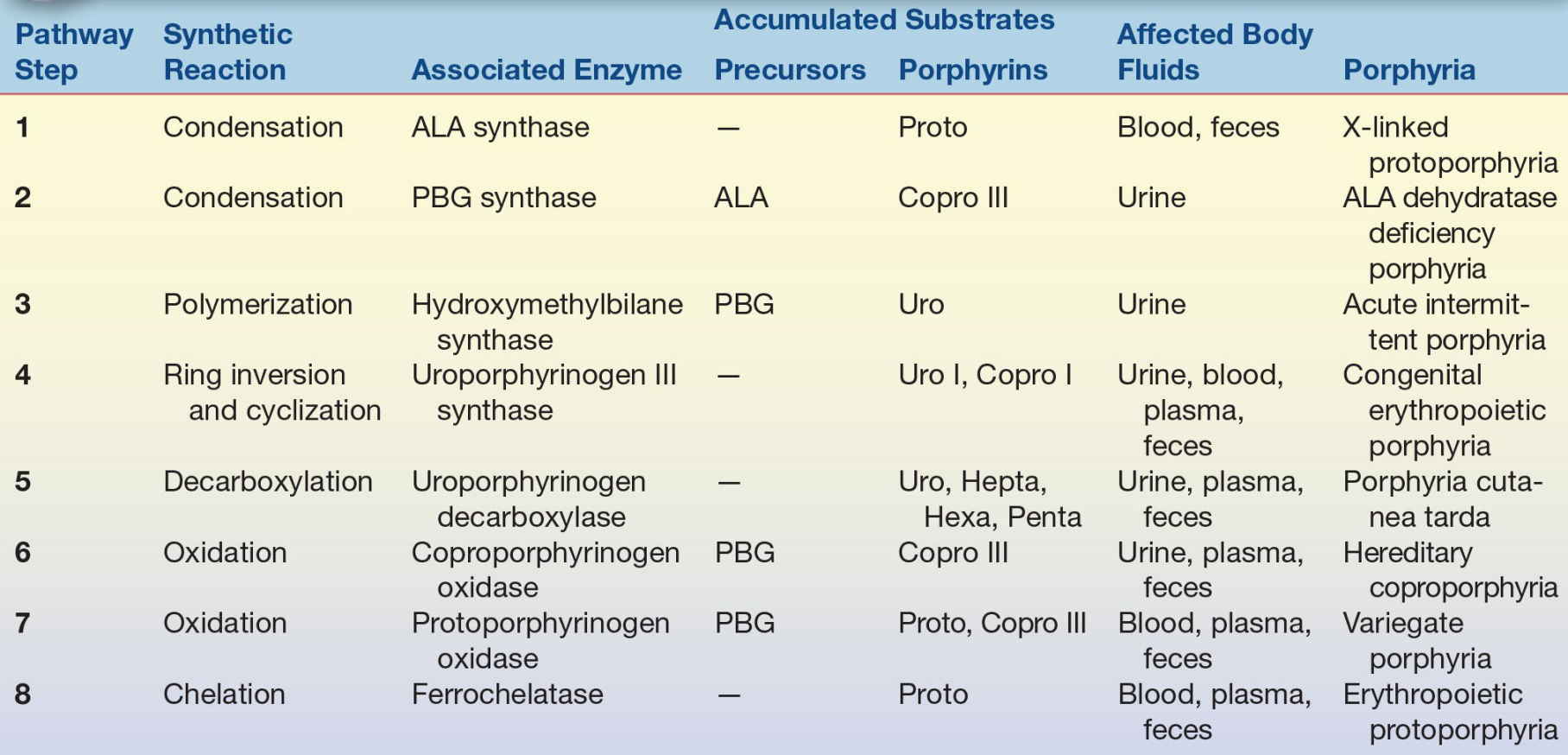
ADP, ALA dehydratase deficiency porphyria; AIP, acute intermittent porphyria; ALA, δ-aminolevulinic acid; CEP, congenital erythropoietic porphyria; Copro, coproporphyrin(ogen); EPP, erythropoietic protoporphyria; HCP, hereditary coproporphyria; Hepta, heptacarboxyl porphyrin(ogen); Hexa, hexacarboxyl porphyrin(ogen); PBG, porphobilinogen; PCT, porphyria cutanea tarda; Penta, pentacarboxyl porphyrin(ogen); Proto, protoporphyrin(ogen); Uro, uroporphyrin(ogen); VP, variegate porphyria; XLPP, X-linked protoporphyria.
Testing for Porphyrin Disorders
When an acute porphyria is suspected, initial testing is determination of PBG in a urine specimen. If symptoms are present and due to porphyria, increased concentrations of PBG will be detected in a random urine specimen.6 A positive test can be followed by measurement of the deficient enzyme, hydroxymethylbilane synthase (PBG deaminase) to identify AIP, and analysis of fecal porphyrins to differentiate among the acute porphyrias (AIP, VP, and HCP). PBG may not be increased in asymptomatic individuals and negative results do not exclude porphyria completely. Testing can be repeated on a specimen collected over 24 hours or when acute symptoms recur.
Initial testing for cutaneous disease requires measurement of porphyrins in a random or timed urine collection.3 Increased excretion of porphyrins in a characteristic pattern particular for each disease is diagnostic for PCT and CEP. Testing for PBG is negative in these conditions. Suspected EPP or XLPP is evaluated by assessing porphyrins in plasma or whole blood. Plasma testing can be used to monitor PCT, but this specimen is not adequate for diagnosis.
Approach to Testing
Logical testing to investigate symptoms and exclude porphyria is based on the separation of disease symptoms into acute (neurological) and cutaneous categories and allows rapid diagnosis of the more common porphyrias—AIP and PCT—associated with each type.17 Detection of increased concentrations of PBG in a urine specimen indicates an acute porphyria, and PCT symptoms are associated with a characteristic pattern of urinary porphyrin isomer excretion. Negative test results allow exclusion of porphyria as a cause of symptoms, while positive test results may provoke additional testing to enumerate the particular porphyria or condition.
In some areas, urine or plasma is screened to detect total porphyrin concentration, and additional (reflex) testing is performed on specimens determined to contain elevated amounts of porphyrins. Since this practice may delay diagnosis, it is not recommended if specimens must be submitted to a remote reference laboratory for testing. Genetic analysis to determine the causative mutation should be limited to individuals with positive biochemical test results that establish the diagnosis of a particular porphyria and family members at risk for disease.10,18
Analytical Methods
Testing for the precursor compounds, PBG and ALA, utilizes ion exchange chromatography to remove interferences and isolate each compound. Detection is accomplished by the addition of 4-dimethylaminobenzaldehyde (Ehrlich’s reagent) to produce a characteristic rose red color that is detected spectrophotometrically. The principle is that of the Watson-Schwartz assay.19 The spectrum has an absorbance maximum at approximately 555 nm and a shoulder at 525 nm. Current assays allow quantitative measurement of analyte concentrations.
Porphyrin intermediates may be evaluated using liquid chromatography (HPLC) with fluorescent detection.3 Urine is acidified to enhance the natural fluorescence of the porphyrins, and an aliquot is injected onto the HPLC system. Separation of the carboxylated porphyrin isomers helps to characterize the particular porphyria. The porphyrin isomers Uro I, Uro III, heptacarboxyl (Hepta), hexacarboxyl, pentacarboxyl, Copro I, and Copro III can be separated within 10 to 12 minutes using a reverse phase analytical column and gradient elution. A dicarboxylated porphyrin (deuteroporphyrin) is used as an internal standard for the assay since these compounds are not found naturally in urine. Measured porphyrin concentrations can be normalized (expressed as a ratio) to creatinine concentration for random urine collections. Results for 24-hour collections are expressed as excretion per day.
Chromatographic analysis of a negative urine specimen shows small amounts of uroporphyrin I and III, little heptacarboxyl porphyrin, more Copro than Uro, and more Copro III than Copro I. Chromatography for a urine specimen positive for PCT reveals large peaks of Uro I, Uro III and Hepta, and Hexa and Penta peaks. Isocoproporphyrin, an alternate metabolic product that is produced when uroporphyrinogen decarboxylase is deficient, is identified in specimens when excretion is high. Massive peaks corresponding to Uro I and Copro I, the isomers that are formed when Uro III synthase is deficient, will be found in a chromatogram characteristic of CEP.
Fecal porphyrin analysis using HPLC with fluorescent detection allows separation and quantification of coproporphyrin isomers and protoporphyrin to differentiate the acute porphyrias AIP, HCP, and VP. An aliquot of the fecal specimen is lyophilized and reconstituted in acidic solution to enhance the natural porphyrin fluorescence before it is subjected to chromatographic analysis. Copro is significantly increased in HCP, and both Proto and Copro may be elevated in VP. Fecal porphyrin excretion is within normal limits (negative) or only slightly increased in AIP. Testing for protoporphyrin to determine presence of EPP or XLPP is performed by scanning fluorescence of a plasma specimen and by analysis of whole blood.
Specimen Requirements
Specimens must be protected from light. Porphyrins and precursor compounds are stable in unpreserved urine at 4°C for up to 48 hours and can be frozen (−20°C) for several weeks. Dilute urine (creatinine <25 mg/dL) is not adequate for analysis. Porphyrins in fecal specimens can be stored frozen (−20°C) for several months. Whole blood collected for protoporphyrin measurement should be anticoagulated with EDTA and stored in the dark at 4°C.3
Since porphyrin analysis typically is offered by large reference laboratories or “porphyrin centers,” the most important considerations may be appropriate selection of the test to perform and careful collection, storage, and transport of the specimen.
HEMOGLOBIN
Role in the Body
Hemoglobin has many important functions in the body. Its major role is oxygen transport to the tissue and CO2 transport back to the lungs. The hemoglobin molecule is designed to take up oxygen in areas of high oxygen tension and release oxygen in areas of low oxygen tension. Hemoglobin is carried to all tissues of the body by erythrocytes. Hemoglobin is also one of the major buffering systems of the body.
Structure of Hemoglobin
The hemoglobin protein is a large, complex molecule that is roughly spherical in shape and is comprised of two major parts: a central oxygen binding, nonprotein heme molecule, and two pairs of identical globin chains (Fig. 19.3). The amino acid sequences of the globin proteins vary and are the basis of fetal/adult chain nomenclature: α, β, δ, and γ.
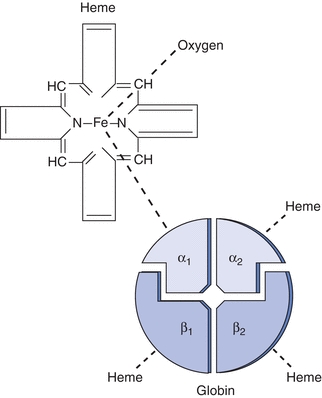
Figure 19.3 Hemoglobin A: structure of the hemoglobin molecule.
The majority of hemoglobin in normal adults is designated as hemoglobin A, or A1, which contains two α and two β chains (Fig. 19.3). Hemoglobin A2, which comprises two α and two δ chains, makes up less than 3% of normal adult hemoglobin. The remainder is composed of hemoglobin F, which contains two α and two γ chains. Two types of γ chains may be present in hemoglobin F, γG and γA. These two types of chains are functionally identical but differ in the amino acid present at the 136th position, glycine or alanine, respectively. Hemoglobin F is the main hemoglobin during fetal life and is about 60% of normal hemoglobin at birth. There is a gradual switch from production of γ chains to β chains, and by about age 9 months, hemoglobin F usually constitutes less than 1% of total hemoglobin. Hemoglobin F has a greater affinity for oxygen than hemoglobin A; therefore, it is a more efficient oxygen carrier for the fetus. Hemoglobin F is more resistant to alkali than hemoglobin A, and this is the basis of one laboratory test to differentiate these two types of hemoglobin.
Two other hemoglobin chains, designated ζ and ε, are present only in embryonic life. Production of these chains stops by week 8 of gestation, and γ chain production takes over. The three embryonic hemoglobins are identified as Gower I, two ζ chains and two ε chains; Gower II, two α chains and two ε chains; and Portland I, two ζ chains and two γ chains.
Genetic control of hemoglobin synthesis occurs in two areas: control of structure and control of rate and quantity of production. Defects in structure produce a group of diseases called the hemoglobinopathies. Defects in rate and quantity of production lead to disorders called the thalassemias. Structurally, each globin chain has its own genetic locus; therefore, it is the individual chains, not the whole hemoglobin molecule, that are under genetic control. The genes for the globin chains can be divided into two major groups: the α genes, located on chromosome 16, and the non–α genes, located on chromosome 11 (Fig. 19.4). In most persons, the α gene locus is duplicated—there are two α chain genes per haploid set of chromosomes, designated α1 and α2. The α gene and, hence, its polypeptide chains are identical in hemoglobins A, A2, and F. The non–α genes for the β, δ, and γ chains are sufficiently close in genetic terms to be subjected to nonhomologous crossover, with the resulting production of fused or hybrid globin chains, such as hemoglobin Lepore (δβ-globin chain) and Kenya (γβ-globin chain).
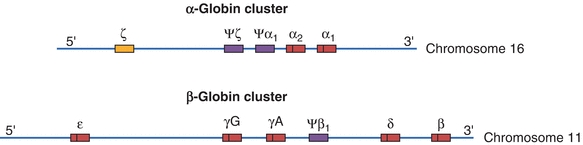
Figure 19.4 Chromosomal structure of the α- and β-globin gene clusters on chromosomes 16 and 11, respectively. Genes with a Ψ (psi) designation represent pseudo-genes. (Used with permission from themedicalbiochemistrypage.org, LLC.)
Based on the genetics of the globin chain production, the structural abnormalities, or hemoglobinopathies, can be divided into four groups:
1. Amino acid substitutions (e.g., hemoglobins S, C, D, E, O, and G)
2. Amino acid deletion—deletions of three or multiples of three nucleotides in deoxyribonucleic acid (DNA; e.g., hemoglobin Gun Hill)
3. Elongated globin chains resulting from chain termination, frame shift, or other mutations (e.g., hemoglobin Constant Spring)
4. Fused or hybrid chains resulting from nonhomologous crossover (e.g., hemoglobins Lepore and Kenya)
The amino acid substitutions are the most common abnormalities, with several hundred described so far. Approximately two-thirds of the hemoglobinopathies have an affected β chain. They may be clinically silent or they may cause severe damage, as with hemoglobin S. Amino acid substitutions, for the majority of defects, result from a single-base nucleotide substitution in DNA.
Absent or diminished synthesis of one of the polypeptide chains of human hemoglobin characterizes the thalassemias, a heterogeneous group of inherited disorders. In α-thalassemia, α-globin chain synthesis is absent or reduced; in β-thalassemia, β-globin chain synthesis is absent (β0-thal) or partially reduced (β+-thal).
Synthesis and Degradation of Hemoglobin
Hemoglobin synthesis occurs in the immature red blood cells (RBCs) located in the bone marrow. Normal synthesis depends on heme synthesis in the cell mitochondria, globin synthesis in the cytoplasm, and on an adequate iron supply. Newly synthesized heme exits the mitochondria and complexes with four globin molecules in the cell cytoplasm. Iron is transported from stores to the developing RBCs where it is inserted into heme and is available to bind oxygen.
Hemoglobin is degraded either in the blood vessels (intravascular hemolysis) or outside of the circulatory system within the phagocytic cells of the spleen, liver, and bone marrow (extravascular hemolysis). Laboratory measurement of hemoglobin degradation products can help to determine increased RBC destruction, such as in a hemolytic anemia (Chapters 11 and 27).
Clinical Significance and Disease Correlation
Hemoglobin Qualitative Defects: The Hemoglobinopathies
Hemoglobin S
The amino acid defect in hemoglobin S is at the sixth position on the β chain, where glutamic acid is substituted by valine, giving the hemoglobin a less negative charge than hemoglobin A. This is the most common hemoglobinopathy in the United States.
CASE STUDY 19.3
A 32-year-old African American woman came to the obstetrics and gynecology clinic of her local hospital because she was feeling a little weak. A CBC showed hemoglobin of 9.9 g/dL (reference range 11.7 to 15.7 g/dL), with an MCV of 87 fL (reference range 80 to 100 fL). The physician ordered a hemoglobin electrophoresis as a follow-up. The cellulose acetate pattern showed a peak of 58% at the hemoglobin A position, a peak of 35% at the hemoglobin S position, and a peak of 5% at the A2 position. Further studies indicated a positive dithionite solubility test result and a hemoglobin F value of 1%.
questions
1. What is the best possible diagnosis for this woman?
2. Does this condition require further follow-up and treatment?
3. What implications does this disease have for her unborn child?
4. Are the values for hemoglobins A2 and F normal for this condition?
Individuals have either sickle cell trait (HbAS, the heterozygous state) or sickle cell disease (HbSS, the homozygous state). Black Africans and African Americans have the highest incidence: 1 of 500 infants has sickle cell anemia and 8% to 10% carry the HbAS trait. It is also found in Mediterranean countries, such as Greece, Italy, and Israel, as well as in Saudi Arabia and India.
Because of the high morbidity and mortality associated with homozygous expression of the gene, the frequency of the mutant gene would be expected to decline in the gene pool. However, a phenomenon known as balanced polymorphism exists, which indicates that the heterozygous state (HbAS) has a selective advantage over either of the homozygous states (HbAA or HbSS). It appears that the heterozygous condition offers protection from parasites, particularly Plasmodium falciparum, especially in children. When infected with P. falciparum, children with sickle cell trait have a lower parasite count, the infection is shorter in duration, and the incidence of death is low. It is thought that the infected RBCs are preferentially sickled and, therefore, efficiently destroyed by phagocytic cells.20
When hemoglobin S is deoxygenated in vitro under near-physiologic conditions, it becomes relatively insoluble as compared with hemoglobin A and aggregates into long, rigid polymers called tactoids. These cells appear as sickle- or crescent-shaped forms on stained blood films. Sickled cells may return to their original shape when oxygenated; however, after several sickling episodes, irreversible membrane damage occurs and cells are phagocytized by macrophages in the spleen, liver, or bone marrow, causing anemia. The severity of the hemolytic process is directly related to the number of damaged cells in circulation. The rigid sickled cells are unable to deform and circulate through small capillaries, resulting in blockage. Tissue hypoxia results, causing extreme pain and leading to tissue death. Infarctions in the spleen are common, causing excessive necrosis and scarring, leading to a nonfunctional spleen in most adults with sickle cell anemia. This is referred to as autosplenectomy. The amount of sickling is related to the amount of hemoglobin S in the cells. The reported inhibitory effect of hemoglobins A and F is due to a dilutional effect. There is also a lower tendency for hemoglobin F to copolymerize with hemoglobin S than with hemoglobin A. This is considered responsible for the observed protective effect of elevated hemoglobin F levels in individuals with sickle cell anemia.
Laboratory findings in the homozygous disease include a normocytic, normochromic anemia, increased reticulocyte count, and variation in size and shape of RBCs with target cells and sickle cells present. Polychromatophilia and nucleated RBCs are common. The heterozygous disease is clinically asymptomatic and usually has a normal blood film. The solubility test for hemoglobin S will be positive in both homozygous and heterozygous forms but should always be confirmed with hemoglobin electrophoresis. On cellulose acetate electrophoresis at an alkaline pH, hemoglobin S moves in a position between hemoglobin A and A2. Of total hemoglobin, 85% to 100% will be hemoglobin S in the homozygous state and usually less than 50% in the heterozygous state. Hemoglobins D and G migrate in the same position as hemoglobin S, but both would be negative with the solubility test. Electrophoresis on citrate agar at an acid pH is necessary to separate these hemoglobins from hemoglobin S (see Fig. 19.4.1).
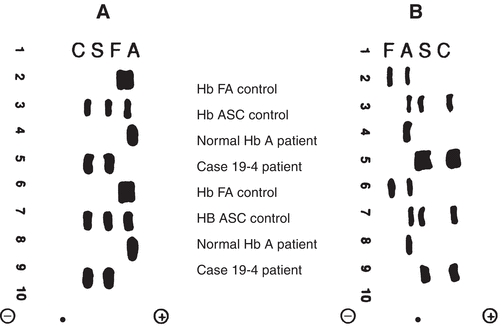
Figure 19.4.1 Hemoglobin electrophoretic patterns. A. Cellulose acetate at pH 8.4. B. Citrate agar at pH 6.2. ● = origin. (Case study data courtesy of Margaret Uthman, MD, University of Texas Medical School at Houston.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


