FIGURE 11.1 General structure of an α-amino acid.
TABLE 11.1 Amino Acids Required in the Synthesis of Proteins
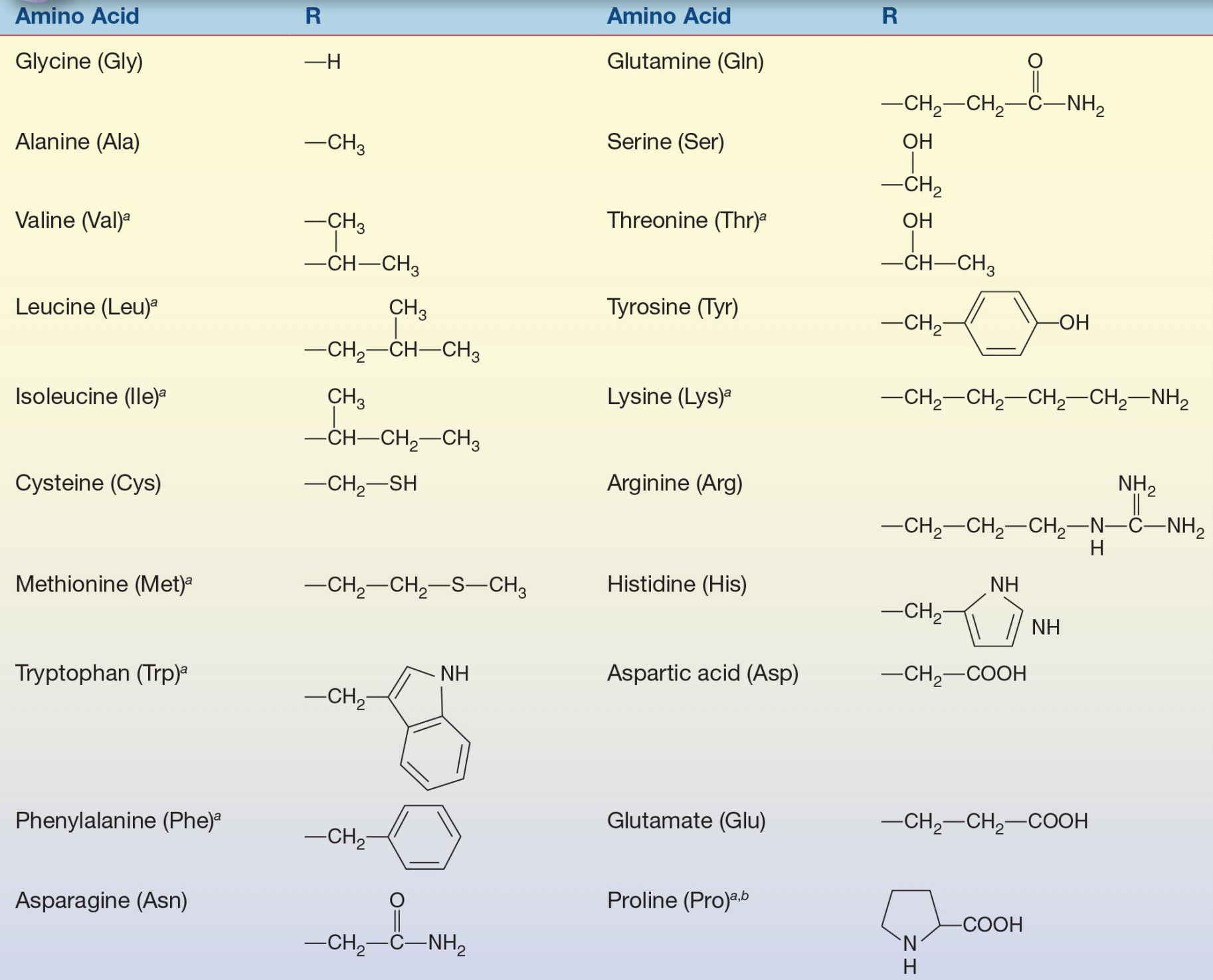
The R group is the group attached to the α carbon.
a Nutritionally essential.
b Exception to attachment of R group is proline.
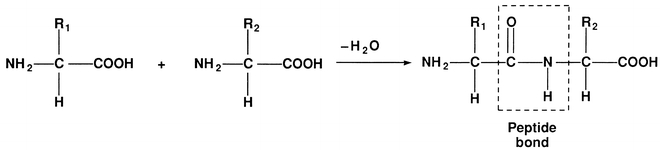
FIGURE 11.2 Formation of a dipeptide.
Metabolism
About half of the 20 amino acids required by humans for protein synthesis cannot be synthesized in vivo at a rapid enough rate to support growth. These nutritionally essential amino acids must be supplied by the diet in the form of proteins. Under normal circumstances, proteolytic enzymes, such as pepsin and trypsin, completely digest dietary proteins into their constituent amino acids. The amino acids are then rapidly absorbed from the intestine into the bloodstream and become part of the body’s pool of amino acids. Amino acids are also released by catabolism of body proteins to be used for energy or synthesis of new proteins.
The essential amino acids are arginine, histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, and valine. Arginine may also be classified as semiessential as dietary supplementation is required in youth, but adults can synthesize adequate amounts to meet the body’s needs. The human body can synthesize sufficient amounts of alanine, asparagine, aspartic acid, cysteine, glutamic acid, glutamine, glycine, proline, serine, and tyrosine. Because additional dietary intake of these amino acids is unnecessary, they are classified as nonessential amino acids. However, some nonessential amino acids are produced from essential ones and a deficiency in the essential amino acid may require supplementation of the nonessential amino acid. For example, tyrosine, a nonessential amino acid, is produced from phenylalanine, an essential amino acid. If the diet is deficient in phenylalanine, tyrosine will also be deficient and may require additional dietary intake.
Amino acids are primarily used in the synthesis of body proteins such as plasma, intracellular, and structural proteins; however, they are also used in the synthesis of nonprotein nitrogen-containing compounds such as purines, pyrimidines, porphyrins, creatine, histamine, thyroxine, epinephrine, and coenzyme NAD. In addition, proteins provide up to 20% of the total energy required daily by the body. To be utilized as an energy source, proteins must first be broken down into their constituent amino acids. The amino group is then removed from the amino acid by either deamination or transamination. The ammonium ion that is produced during deamination of the amino acids is converted into urea via the urea cycle in the liver.2 The resultant ketoacid enters into a common metabolic pathway with carbohydrates and fats to be converted into useable energy. Glucogenic amino acids generate precursors of glucose, such as pyruvate or citric acid cycle intermediates. Examples include alanine, which can be deaminated to pyruvate; arginine, which is converted to α-ketoglutarate; and aspartic acid, which is converted to oxaloacetate. Ketogenic amino acids, such as leucine and lysine, are degraded to acetyl-CoA or acetoacetyl-CoA and form ketone bodies. Isoleucine, phenylalanine, tryptophan, tyrosine, and threonine are both ketogenic and glucogenic.
Essential Amino Acids
Arginine (Arg)
Arginine is a complex amino acid often found at the catalytic (active) site in proteins and enzymes due to its amine-containing side chain. Arginine plays an important role in cell division, wound healing, stimulation of protein synthesis, immune function, and the release of hormones. One important role of arginine is the conversion of ammonia, which is highly toxic, into urea, which can be safely excreted via the urinary system.
Histidine (His)
Histidine is one of the basic by pH amino acids due to its imidazole side chain. Histidine is needed to help grow and repair body tissues and to maintain the myelin sheaths that protect nerve cells and is the direct precursor of histamine, which is involved in immune response. It also plays an important role in the manufacturing of red and white blood cells and in protecting the body from heavy metal toxicity and is an important source of carbon atoms in the synthesis of purines for DNA and RNA synthesis.
Isoleucine (Ile), Leucine (Leu), and Valine (Val)
Isoleucine, leucine, and valine are branched-chain amino acids that are collectively referred to as the branched-chain amino acid group. These branched-chain amino acids promote healing of muscle tissue, skin, and bones; help regulate blood glucose concentrations; and maintain energy levels. Additionally, isoleucine plays an important role in hemoglobin formation, whereas leucine is necessary for optimal growth in infants. Leucine and valine also aid in maintaining the nitrogen balance in adults. Valine is used in treatments for muscle, mental, and emotional problems such as insomnia and anxiety, as well as liver and gallbladder disease.
Lysine (Lys)
Lysine has a net positive charge, which makes it one of three basic, by charge, amino acids. It plays a role in the production of antibodies and lowering triglyceride levels. Lysine also helps in the absorption and conservation of calcium and plays an important role in the formation of collagen, a component of cartilage and connective tissue.
Methionine (Met)
Methionine is an important amino acid that helps to initiate translation of messenger RNA (mRNA) by being the first amino acid incorporated into the N-terminal position of all proteins.3 Methionine is a source of sulfur, which is required for normal metabolism and growth. Methionine also assists in the breakdown of fats, helps to detoxify lead and other heavy metals, helps diminish muscle weakness, and prevents brittle hair. Methionine reacts with adenosine triphosphate (ATP) in the synthesis of many important substances, including epinephrine and choline.
Phenylalanine (Phe)
Phenylalanine is classified as a nonpolar amino acid because of the hydrophobic nature of its benzyl side chain. It promotes alertness and vitality, elevates mood, decreases pain, aids in memory and learning, and is used to treat arthritis and depression. Phenylalanine uses an active transport channel to cross the blood–brain barrier and is used by the brain to produce norepinephrine, a neurotransmitter that transmits signals between nerve cells. In large quantities, phenylalanine can interfere with production of serotonin, another neurotransmitter. Phenylalanine is the direct metabolic precursor of tyrosine, and deficiency of phenylalanine will also result in a deficiency of tyrosine.
Threonine (Thr)
Threonine is an alcohol-containing amino acid that is an important component in the formation of collagen, elastin (a connective tissue protein), and tooth enamel. It is also important in the production of neurotransmitters and health of the nervous system. Threonine helps maintain proper protein balance in the body and aids liver function, metabolism, and assimilation.
Tryptophan (Trp)
Tryptophan is a metabolic precursor for serotonin, melatonin (a neurohormone), and niacin (a vitamin). Tryptophan is a natural relaxant that helps alleviate insomnia by inducing sleep, soothes anxiety, and reduces depression. It is used in the treatment of migraine headaches, aids in weight control by reducing appetite, and helps control hyperactivity in children.
Nonessential Amino Acids
Alanine (Ala)
Alanine is one of the simplest amino acids and is a product of the breakdown of DNA or the dipeptides, anserine and carnosine. It is also formed as a result of glucose metabolism (glycolysis) in muscle tissue and the conversion of pyruvate, a pivotal compound in carbohydrate metabolism.4 Alanine plays a major role in the transfer of nitrogen from peripheral tissues to the liver for processing and excretion and strengthens the immune system through production of antibodies. Alanine also helps in reducing the buildup of toxic substances released when muscle protein is broken down quickly to meet the energy demands of the body.
Asparagine (Asn)
Asparagine is derived from aspartic acid and ATP through transamidation, which is the transfer of an amide group from one compound to another.4 Asparagine is one of the principal and most abundant amino acids involved in the transport of nitrogen. It is required by the nervous system and plays an important role in the synthesis of ammonia; however, a primary function of asparagine is in the conversion of one amino acid into another via amination or transamination. Amination is the process by which an amine group is introduced into an organic molecule, and transamination is the transfer of an amino acid to an α-ketoacid.
Aspartic Acid (Asp)
Aspartic acid, or aspartate, is synthesized from oxaloacetate through transamination. An important role of aspartic acid is as the precursor for several other amino acids. Amino acids that are synthesized from aspartic acid include asparagine, arginine, lysine, methionine, threonine, isoleucine, and several nucleotides. Aspartic acid is a metabolite in the citric acid cycle and the urea cycle. It also participates in the generation of glucose from nonsugar substrates, which is known as gluconeogenesis.
Cysteine (Cys)
Cysteine is classified as a nonessential amino acid; however, infants, the elderly, and individuals with certain metabolic diseases or intestinal absorption issues may require supplementation. Cysteine should not be confused with cystine as they are two different amino acids. However, cysteine is named after cystine as it is an oxidized dimer of cysteine.5 Cysteine is potentially toxic, so it is absorbed during digestion as cystine, which is more stable in the gastrointestinal tract and less toxic. Cystine is transported to cells, where it is reduced to two cysteine molecules upon cell entry. Cysteine can also be synthesized in vivo from the essential amino acid methionine through a series of enzymatic reactions. Cysteine is an important structural and functional component of many proteins and enzymes and has significant antioxidant properties.
Glutamic Acid (Glu)
Glutamic acid, or glutamate, is synthesized from the transamination of a number of amino acids such as alanine and aspartic acid. When an amino group is added to glutamic acid, it forms another important amino acid, glutamine. Glutamic acid is one of two amino acids that have a net negative charge by pH, making it a very polar molecule. Glutamic acid serves as a neurotransmitter, and its dysregulation has been linked to epileptic seizures. It is also important in the metabolism of sugars and fats and aids in the transport of potassium into spinal fluid.
Glutamine (Gln)
Glutamine is the most abundant amino acid in the body and is synthesized from glutamic acid. Glutamine has many important functions including renal maintenance of the acid–base balance, providing fuel for a healthy digestive tract,6 and acting as the basis of the building blocks for synthesis of RNA and DNA. Glutamine is also a source of cellular energy and aids in immune function. Another vital role of glutamine is the transport of ammonia to the liver for metabolism. Ammonia is a by-product of protein breakdown. It is transported to the liver where it is converted into urea and excreted by the kidneys. Studies have shown glutamine to be useful in the treatment of serious illnesses, injury, trauma, burns, cancer treatment–related side effects, and wound healing for postoperative patients.7
Glycine (Gly)
Glycine is synthesized from another nonessential amino acid, serine. Glycine is essential for synthesis of nucleic acids, bile acids, proteins, peptides, purines, ATP, porphyrins, hemoglobin, glutathione, creatine, bile salts, glucose, glycogen, and other amino acids. The liver utilizes glycine in the detoxification of compounds and in the synthesis of bile acids. Glycine is also an inhibitory neurotransmitter in the central nervous system (CNS) and a metal complexing agent. Additionally, glycine retards muscle degeneration, improves glycogen storage, and promotes healing.
Proline (Pro)
Proline is produced from glutamine and is the precursor for hydroxyproline. Hydroxyproline is manufactured into collagen, tendons, ligaments, and cardiac tissue. Proline is involved in wound healing, especially that of cartilage, and in the strengthening of joints, tendons, and cardiac tissue. It also works in tandem with vitamin C to promote healthy connective tissues.
Serine (Ser)
Serine is synthesized from 3-phosphoglycerate, which is an intermediate in glycolysis. Serine is needed for the proper metabolism of lipids and fatty acids and plays an important role in the body’s synthetic pathways for pyrimidines, purines, creatine, and porphyrins. It is highly concentrated in all cell membranes, is a component of the protective myelin sheaths surrounding nerve fibers, and aids in the production of antibodies for the maintenance of a healthy immune system.
Tyrosine (Tyr)
Tyrosine is metabolically synthesized from phenylalanine. Tyrosine is a precursor of the adrenal hormones epinephrine, norepinephrine, and dopamine and thyroid hormones including thyroxine. It is important in overall metabolism and aiding in function of the adrenal glands, thyroid, and pituitary glands. Tyrosine stimulates metabolism and the nervous system and acts as a mood elevator, making it useful in the treatment of chronic fatigue, narcolepsy, anxiety, depression, low sex drive, allergies, and headaches.
Recently Identified Amino Acids
Selenocysteine (Sec)
Selenocysteine is recognized as the 21st amino acid, but unlike other amino acids present in proteins, it is not coded for directly in the genetic code. Selenocysteine is encoded by a UGA codon, which is normally a stop codon; however, like the other amino acids used by cells, selenocysteine has a specialized transfer RNA (tRNA). Selenocysteine was named as an amino acid in 2002 and was found to be the selenium analogue of cysteine, in which a selenium atom replaces sulfur. Selenocysteine is present in several enzymes, such as formate dehydrogenases, glycine reductases, and some hydrogenases. It has been discovered that HIV-1 encodes a functional selenoprotein, and patients with HIV infection have been shown to have a lower-than-average blood plasma selenium level.8,9
Pyrrolysine (Pyl)
Pyrrolysine is the 22nd naturally occurring genetically encoded amino acid used by some prokaryotes and single-celled microorganisms in enzymes that are part of their methane-producing metabolism. This lysine derivative is encoded by the UAG codon, normally a stop codon, possibly modified by the presence of a specific downstream sequence forcing the incorporation of pyrrolysine instead of terminating translation.10 This amino acid is not present in humans.
Aminoacidopathies
Aminoacidopathies are a class of inherited errors of metabolism in which there is an enzyme defect that inhibits the body’s ability to metabolize certain amino acids. The abnormalities exist either in the activity of a specific enzyme in the metabolic pathway or in the membrane transport system for the amino acids. Aminoacidopathies cause severe medical complications, such as brain damage, due to the accumulation of toxic amino acids or their by-products in the blood and tissues. Due to the severity of these complications, many states require newborn screening tests to aid in early diagnosis and initiation of therapies. Phenylketonuria (PKU) is one of the most well-known aminoacidopathies and was the first newborn screening test introduced in the early 1960s. Now, some states require screening tests for up to 26 amino acids,11 and more than 100 diseases have been identified that result from inherited errors of amino acid metabolism.
Phenylketonuria
PKU is inherited as an autosomal recessive trait and occurs in about 1 in 15,000 births in the United States. The metabolic defect in the classic form of PKU is an absence of activity of the enzyme phenylalanine hydroxylase (PAH) due to mutations in the PAH gene. PAH catalyzes the conversion of phenylalanine to tyrosine as illustrated in Figure 11.3. In the newborn, the upper limit of normal for phenylalanine concentration in the blood is 120 μmol/L (2 mg/dL).12 In the absence of PAH activity, phenylalanine concentrations are usually greater than 1,200 μmol/L and in untreated classic PKU, blood concentrations as high as 2.4 mmol/L can be found. Chronically elevated blood concentrations of phenylalanine and its metabolites, such as phenylpyruvic acid, phenylpyruvate (also known as phenylketone), and phenyllactic acid, can cause significant and permanent brain damage.
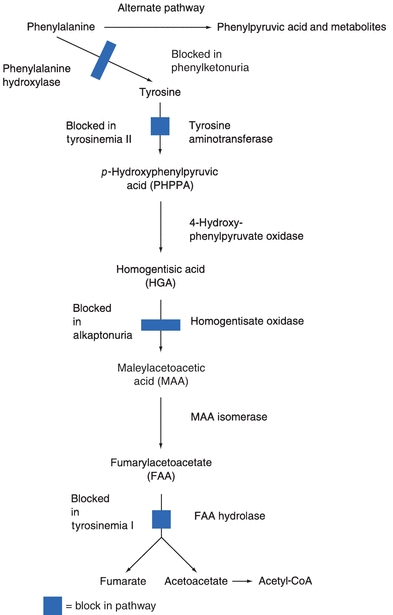
FIGURE 11.3 Metabolism of phenylalanine and tyrosine.
Phenylalanine and its metabolites are found in both the blood and urine of patients with PKU and cause the urine to have a characteristic “musty” or “mousy” odor. Partial deficiencies of PAH activity are typically classified as mild PKU and result in phenylalanine concentrations between 600 to 1,200 μmol/L. Patients with phenylalanine concentrations in the range of 180 to 600 μmol/L with no accompanying accumulation of phenylketones are classified as having non-PKU mild hyperphenylalaninemia.
In infants and children with this inherited defect, retarded mental development and microcephaly occur as a result of the toxic effects of phenylalanine or its metabolic by-products on the brain. Brain damage can be avoided if the disease is detected at birth and the infant is maintained on a diet containing very low levels of phenylalanine. Women with untreated PKU during pregnancy almost always have infants who are microcephalic and mentally retarded. However, fetal effects of maternal PKU are preventable if the mother maintains a phenylalanine-restricted diet before conception and throughout pregnancy.
Hyperphenylalaninemia can also be the result of a deficiency in the enzymes needed for regeneration and synthesis of tetrahydrobiopterin (BH4). BH4 is a cofactor required for the enzymatic hydroxylation of phenylalanine, tyrosine, and tryptophan. Deficiency of BH4 results in elevated blood concentrations of phenylalanine and deficient production of neurotransmitters from tyrosine and tryptophan. Although cofactor defects account for only 1% to 5% of all cases of hyperphenylalaninemia, they must be differentiated from PKU so that appropriate treatment with the active cofactor along with the neurotransmitter precursors l-DOPA and 5-OH tryptophan can be initiated.13
Screening for PKU is now required in all states in the United States, and blood is collected at about 3 days of age. If the screening test is abnormal, other tests are performed to confirm or exclude PKU. Once diagnosed as PKU, the goal of treatment is to maintain the blood concentration of phenylalanine between 120 and 600 μmol/L (2 and 10 mg/dL) through diet modification. Phenylalanine is still required for normal growth; therefore, a diet with some phenylalanine is recommended, but in much lower amounts than a typical diet contains. High-protein foods, such as meat, fish, poultry, eggs, cheese, and milk, should be avoided, and calculated amounts of cereals, starches, fruits, and vegetables, along with a milk substitute, are usually recommended.
In 2007, the U.S. Food and Drug Administration (FDA) approved sapropterin dihydrochloride (Kuvan®), the first drug to help manage PKU.14 The drug helps reduce phenylalanine concentrations by increasing the activity of the PAH enzyme. This drug is only effective for patients that have some PAH activity, who continue to follow a phenylalanine-restricted diet and have their phenylalanine levels monitored.
The Guthrie test is a semiquantitative, bacterial inhibition assay for phenylalanine based on the ability of phenylalanine to facilitate bacterial growth in a culture medium despite the presence of a growth inhibitor. To perform the Guthrie test, an agar gel plate containing β-2-thienylalanine, a growth inhibitor, is inoculated with Bacillus subtilis. Blood is collected from the newborn onto a piece of filter paper, and a small disk is punched out of the filter paper and placed on the inoculated agar plate. The β-2-thienylalanine present in the agar inhibits bacterial growth; however, in the presence of phenylalanine leached from the impregnated filter paper disk, inhibition is overcome and bacteria grow around the disk. The Guthrie test is sensitive enough to detect serum phenylalanine concentrations of 180 μmol/L (3 mg/dL). The test has been widely used throughout North America and Europe as one of the core newborn screening tests since the late 1960s. In recent years, this test has been replaced in many areas by newer techniques, such as high-performance liquid chromatography (HPLC) and tandem mass spectrometry (MS/MS).
Another screening test for PKU involves a microfluorometric assay for the direct measurement of phenylalanine in dried blood filter disks. In contrast to the Guthrie test, this method yields quantitative results, is more adaptable to automation, and is not affected by the presence of antibiotics. The procedure is based on the fluorescence of a complex formed by phenylalanine, ninhydrin, and copper in the presence of a dipeptide. The test requires pretreatment of the dried blood filter disk with trichloroacetic acid (TCA). The extract is then added to a microtiter plate with a mixture of ninhydrin, succinate, and leucylalanine in the presence of copper tartrate. The fluorescence of the complex is measured using excitation/emission wavelengths of 360 and 530 nm, respectively.15
Positive screening results for PKU must be verified by another method. One rapid diagnostic procedure for neonatal PKU uses microwave-assisted silylation followed by gas chromatography–mass spectrometry (GC/MS). In this procedure, amino acids are extracted from neonatal blood samples, rapidly derived with N,O–bis(trimethylsilyl)-trifluoroacetamide under microwave irradiation, and the derivatives are analyzed by GC/MS.
The reference method for quantitative serum phenylalanine is HPLC; however, MS/MS is now considered to be the gold standard for detecting a variety of congenital diseases in newborns. Mass spectrometry is an analytical technique that measures the mass-to-charge ratio of charged particles.16 It is most commonly used to identify the composition of a specimen by generating a mass spectrum representing the masses of the specimen’s components. Patients with PKU generally demonstrate an increase in phenylalanine concentrations and a decrease in tyrosine concentrations. Using MS/MS, both analytes can be quantitated, and a ratio of phenylalanine to tyrosine (Phe/Tyr) can be calculated. Using the ratio between the analytes rather than individual concentrations increases the specificity of the measurement and lowers the false-positive rate for PKU to less than 0.01%. MS/MS methods have greater sensitivity allowing detection of lower concentrations of phenylalanine and earlier diagnosis of PKU, sometimes as early as the first day of life. Most newborn screening MS/MS methods have the ability to detect more than 25 different genetic disorders with a single specimen so this technology is rapidly replacing the multiple procedures currently used in newborn screening programs when the laboratory can support MS/MS testing.
Prenatal diagnosis and detection of carrier status in families with PKU are now available using DNA analysis as PKU results from multiple independent mutations (>400 identified) at the PAH locus.
Tyrosinemia
Tyrosinemia is another inherited autosomal recessive aminoacidopathy. This inborn metabolic disorder of tyrosine catabolism is characterized by the excretion of tyrosine and its catabolites in urine or tyrosinuria. There are three types of tyrosinemia each caused by deficiencies of different enzymes in the metabolic pathway of tyrosine.
Type I tyrosinemia is the most severe form occurring in about 1 in 100,000 births worldwide. This form of tyrosinemia is caused by a mutation in the FAH gene, which codes for the enzyme fumarylacetoacetate hydrolase (FAH). FAH is the last of five enzymes required for metabolism of tyrosine. Symptoms of type I tyrosinemia generally manifest in the first few months of life and include failure to thrive, diarrhea, vomiting, jaundice, a cabbage-like odor, distended abdomen, swelling of the legs, and an increased predisposition for bleeding. Type I tyrosinemia can lead to liver and kidney failure, problems affecting the nervous system such as peripheral neuropathy, and an increased risk of liver cancer later in life. Untreated, patients with type I tyrosinemia typically do not live past the age of ten.
Type II tyrosinemia is caused by a mutation in the TAT gene, which leads to a deficiency of the enzyme tyrosine aminotransferase (TAT). Type II tyrosinemia occurs in less than 1 in 250,000 births worldwide. TAT is the first enzyme of the five enzymes required for the conversion of tyrosine to smaller molecules that are excreted by the kidneys or used in energy-producing reactions. About half of the individuals with type II tyrosinemia have impaired mental development. Other symptoms manifest in the eyes and skin including abnormal light sensitivity (photophobia), eye pain and redness, and painful skin lesions on the palms and soles of the feet.
Type III tyrosinemia is a rare disorder with only a few cases having been reported worldwide. It is caused by a mutation in the HPD gene that results in deficiency of the enzyme 4-hydroxyphenylpyruvate dioxygenase (HPD). HPD is the second enzyme of the cascade required for tyrosine metabolism and is found mainly in the liver, with lesser amounts found in the kidneys. Patients with type III tyrosinemia typically have impaired mental development, seizures, and some intermittent loss of balance and coordination.
Diagnostic criteria for tyrosinemia include an elevated tyrosine blood concentration using MS/MS and an elevated concentration of succinylacetone.17 Succinylacetone is a toxic metabolite that forms when tyrosine cannot be metabolized through the appropriate enzymatic pathway. Succinylacetone can cause severe damage to the liver and kidneys resulting in the need for a full or partial liver transplant. The treatment for tyrosinemia is generally a low-protein diet, but may also include medications such as nitisinone (NTBC). Nitisinone prevents the formation of maleylacetoacetic acid and fumarylacetoacetic acid, which can be converted to succinylacetone. Since nitisinone’s first use for treatment of tyrosinemia in 1991, it has replaced liver transplantation as the first-line treatment for this rare condition.
Alkaptonuria
Alkaptonuria is another inherited autosomal recessive amino acid disorder with an occurrence of about 1 in 250,000 births worldwide. A mutation in the HGD gene leads to a deficiency of the enzyme homogentisate oxidase (HGD), which is required for proper metabolism of tyrosine and phenylalanine. As a result of this enzyme deficiency, patients with alkaptonuria have elevated concentrations of homogentisic acid (HGA) in the blood, deposit HGA in connective tissues, and excrete HGA in their urine. Patients with alkaptonuria typically do not present with symptoms until their third decade of life. Over time, HGA is deposited in the cartilage of the ears, nose, and tendons of the extremities and results in a dark blue-black pigmentation known as ochronosis. Small dark spots on the sclera of the eye may also be seen. HGA also accumulates in the cartilage of joints, such as the hips, and causes an arthritis-like degeneration, which is frequently the first symptom reported by patients. HGA is excreted through the renal system and is found in the urine of patients with alkaptonuria. If the urine specimen is allowed to mix with air, it will eventually result in a brownish-black color due to oxidization of HGA.
The ferric chloride test may be performed as a rapid screen for the presence of HGA in urine. When ferric chloride is added to an aliquot of urine that contains HGA, it will turn black due to the interaction with the phenol group. Patients with alkaptonuria may be prescribed high-dose vitamin C supplementation as this vitamin has been shown to slow down the accumulation of HGA in the cartilage, which may slow the progression of arthritis.
Maple Syrup Urine Disease
Maple syrup urine disease (MSUD) results from an absence or greatly reduced activity of a complex of enzymes known as branched-chain α-ketoacid decarboxylase (BCKD). This deficiency is inherited in an autosomal recessive pattern and is the result of a mutation in the BCKDHA, BCKDHB, or DBT genes. Deficiency of the BCKD complex inhibits the normal metabolism of the three essential branched-chain amino acids leucine, isoleucine, and valine. MSUD has been part of several state newborn screening programs since the mid-1970s, with a reported occurrence of 1 in 185,000 births worldwide. The most striking feature of this hereditary disease is the characteristic “maple syrup” or “burnt sugar” odor of the urine, breath, and skin of patients with MSUD. This characteristic odor is the result of the accumulation of the branched-chain amino acids and their corresponding ketoacids in the blood, urine, and cerebrospinal fluid (CSF).
Newborns with MSUD typically develop lethargy, vomiting, lack of appetite, and signs of failure to thrive within a week of birth. Elevated concentrations of leucine, isoleucine, valine, and their associated ketoacids can be toxic to the brain and may result in muscle rigidity, stupor, and respiratory irregularities. Undiagnosed and untreated, the disease can progress to cause severe mental retardation, seizures, acidosis, and hypoglycemia and can lead to death. If diagnosed early on, MSUD can be controlled by restricting dietary proteins to minimize leucine, isoleucine, and valine.
A modified Guthrie test can be used to screen for MSUD. An agar gel plate containing 4-azaleucine, another growth inhibitor, is inoculated with Bacillus subtilis. A small disk is punched out of filter paper that contains blood from the newborn and is placed on the inoculated agar plate. The 4-azaleucine present in the agar inhibits bacterial growth; however, in the presence of the branched-chain amino acids leached from the impregnated filter paper disk, inhibition is overcome and bacteria grow around the disk. A microfluorometric assay for the three branched-chain amino acids uses a filter paper specimen treated with a solvent mixture of methanol and acetone to denature the hemoglobin. Leucine dehydrogenase is added to an aliquot of the extract, and the fluorescence of the NADH produced in the subsequent reaction is measured at 450 nm, with an excitation wavelength of 360 nm.18 A leucine concentration greater than 4 mg/dL is indicative of MSUD. MS/MS is also being used in screening and quantitative testing for MSUD. Prenatal diagnosis of MSUD is made by testing the decarboxylase enzyme concentration in cells cultured from amniotic fluid.
Isovaleric Acidemia
Isovaleric acidemia (IVA) is an autosomal recessive disorder caused by mutation of the IVD gene, which codes for isovaleryl-CoA dehydrogenase (IVD). IVD plays an important role in the metabolism of leucine.
Isovaleric acidemia occurs in approximately 1 in 250,000 births in the United States. Some people with gene mutations that cause isovaleric acidemia are asymptomatic and never experience any signs or symptoms of the condition. For those patients with IVA that are not asymptomatic, a characteristic feature of IVA is a distinctive odor of sweaty feet caused by the buildup of isovaleric acid. Other clinical manifestations of this disorder may become apparent as early as a few days after birth and include failure to thrive, vomiting, and lethargy. Health problems related to IVA range from very mild to life threatening, such as seizures and coma, but, when severe, can result in permanent damage to the brain and nervous system and possible death.
Treatment for IVA includes a protein-restrictive diet to lower the levels of accumulating isovaleric acid, which is toxic to the central nervous system. Oral administration of glycine and carnitine supplementation may be prescribed because they interact with isovaleric acid to form nontoxic, readily excreted products.
If IVA is suspected, the urine of newborns can be screened for isovaleric acid using MS/MS or chromatography. Laboratory results for patients with IVA reveal metabolic acidosis, mild-to-moderate ketonuria, hyperammonemia, thrombocytopenia, and neutropenia.
Homocystinuria
Homocystinuria is yet another inherited autosomal recessive disorder of amino acid metabolism. Mutations in the CBS, MTHFR, MTR, MTRR, and MMADHC can result in homocystinuria. The most common form of homocystinuria is the result of a mutation in the CBS gene and occurs in about 1 in 200,000 births worldwide. This mutation results in a deficiency of the enzyme cystathionine β-synthase, which is necessary for metabolism of methionine. This deficiency results in elevated plasma and urine concentrations of methionine and of the precursor homocysteine. Symptoms associated with this form of homocystinuria include near-sightedness; dislocation of the lens in the eye, caused by the lack of cysteine synthesis essential for collagen formation; osteoporosis; and, frequently, mental retardation.19
Treatment includes dietary restriction of proteins to reduce concentrations of methionine as well as high doses of vitamin B6. Those who do not respond to this treatment may need trimethylglycine and folic acid supplementation.
Newborn screening for homocystinuria may also be performed using a modified Guthrie test. In this form of the test, l-methionine sulfoximine is utilized as the bacterial growth inhibitor and increased plasma methionine levels will result in bacterial growth. HPLC and MS/MS methods are commonly used as confirmatory methods, and a methionine concentration greater than 2 mg/dL indicates homocystinuria. Alternatively, urinary total homocysteine concentrations can be measured using liquid chromatography–tandem mass spectrometry (LC-MS/MS).20
Citrullinemia
Citrullinemia belongs to a class of genetic diseases called urea cycle disorders. In the urea cycle, excess nitrogen, generated during protein metabolism, is used in the formation of urea, which can be safely excreted in urine. Citrullinemia is inherited in an autosomal recessive pattern and occurs in two forms.
Type I citrullinemia is the result of a mutation in the ASS1 gene. This gene codes for synthesis of the enzyme argininosuccinic acid synthetase (ASS). A deficiency of ASS leads to an accumulation of the amino acid citrulline as well as ammonia in the blood due to impaired conversion of ammonia into urea for excretion. Type I citrullinemia is the most common form occurring in about 1 in 57,000 births worldwide. In affected infants, clinical symptoms include lack of appetite, failure to thrive, vomiting, lethargy, seizures, and coma as toxic ammonia builds up in the body. If not treated promptly, severe brain damage or death may occur.
Type II citrullinemia is caused by a mutation in the SLC25A13 gene that codes for production of the transport protein citrin. Citrin helps transport molecules inside the cell that are used in the production and breakdown of simple sugars, production of proteins, and the urea cycle. In type II citrullinemia, cells are prevented from making citrin, which inhibits the urea cycle and disrupts the production of proteins and nucleotides. The resulting accumulation of ammonia and other toxic substances leads to confusion, restlessness, memory loss, personality changes, seizures, and coma.
Treatment of citrullinemia includes a high-caloric, protein-restrictive diet; arginine supplementation, and administration of sodium benzoate and sodium phenylacetate.
Argininosuccinic Aciduria
Argininosuccinic aciduria (ASA) is another urea cycle amino acid disorder that is inherited in an autosomal recessive pattern and is the result of a mutation in the ASL gene. ASA occurs in approximately 1 in 70,000 births worldwide. Infants with argininosuccinic acidemia lack the enzyme argininosuccinic acid lyase (ASL), which plays a role in the conversion of ammonia into urea. When ASL is deficient, the urea cycle cannot proceed normally and nitrogen accumulates in the blood in the form of ammonia. Ammonia is especially damaging to the nervous system and can result in seizures as well as possible coma. Clinical symptoms of ASA typically develop within the first few days of life and begin with lethargy and unwillingness to eat.
Treatment of ASA is similar to that for citrullinemia and includes a high-calorie, protein-restrictive diet; arginine supplementation; and administration of sodium benzoate and sodium phenylacetate. It should be noted that newborn screening tests cannot differentiate citrullinemia from argininosuccinic acidemia.21
Cystinuria
Cystinuria is an inherited autosomal recessive disorder, occurring in approximately 1 in 10,000 births worldwide, which is caused by mutations in the SLC3A1 and SLC7A9 genes. These genes code for the synthesis of a protein complex found in the kidneys that is responsible for the reabsorption of cystine from urine. Cystinuria is characterized by inadequate reabsorption of cystine during urine formation in the kidneys resulting in an elevated concentration of cystine. When saturated, cystine can precipitate out of the urine and form stones in the kidneys, ureters, or bladder. Kidney stones often recur throughout a patient’s lifetime and are responsible for the symptoms of the disease, including hematuria, flank pain, and, frequently, urinary tract infections.
Treatment for cystinuria is generally aimed at preventing the formation of cystine stones. This may be accomplished by increasing the patient’s fluid intake to 4 L/day, which serves to increase the volume of urine, reduce the concentration of cystine, and minimize the likelihood of it precipitating and forming stones. When a high fluid intake does not reduce the formation of stones, penicillamine may be prescribed. Penicillamine forms a more soluble complex with cystine, which prevents it from precipitating and forming stones. Once stones have formed, percutaneous nephrolithotripsy may be performed as an alternative to surgery to remove the kidney stones.
Cystinuria can be diagnosed by testing the urine for cystine using cyanide nitroprusside, which produces a red-purple color on reaction with sulfhydryl groups. False-positive results as a result of homocysteine must be ruled out. Patients with cystinuria also excrete elevated concentrations of lysine, arginine, and ornithine. These amino acids are more soluble in urine and generally do not lead to stone formation; however, ion exchange chromatography can be used for quantitative analysis, which may aid in the diagnosis of cystinuria.
Methods of Analysis
Blood samples for amino acid analysis should be drawn after at least 6 to 8 hours of fasting to avoid the effect of absorbed amino acids originating from dietary proteins. The sample is collected in a heparin tube with the plasma promptly removed from the cells, taking care not to aspirate the platelet and white cell layer to prevent contamination with platelet or leukocyte amino acids. White blood cell levels of aspartic acid and glutamic acid, for example, are about 100 times higher than those in plasma; hemolysis is unacceptable for the same reason. Deproteinization should be performed within 30 minutes of sample collection, and analysis should be performed immediately or the sample frozen at –20°C to –40°C.
Urinary amino acid analysis can be performed on a random specimen for screening purposes. For quantitation, a 24-hour urine specimen preserved with thymol or organic solvents is required. Amniotic fluid may also be analyzed.
For amino acid screening, the method of choice is thin-layer chromatography. The application of either one- or two-dimensional separations depends on the purpose of the analysis. If searching for a particular category of amino acids, such as branched-chain amino acids or a single amino acid, usually, a one-dimensional separation is sufficient. For more general screening, two-dimensional chromatography is essential. The amino acids migrate along one solvent front, and then the chromatogram is rotated 90° and a second solvent migration performed. A variety of solvents have been used, including butanol, acetic acid, water and ethanol, and ammonia and water mixtures. The chromatogram is viewed by staining with ninhydrin, which gives most amino acids a blue color. Amino acids can also be separated and quantitated by ion exchange chromatography, an HPLC reversed-phase system equipped with fluorescence detection,22 or capillary electrophoresis. As discussed, another technique that provides a highly specific and sensitive method for the measurement of amino acids is MS/MS.23
PROTEINS
Overview
Proteins play key roles in every cell throughout the human body. Some of their major functions include catalyzing biochemical reactions as enzymes, transporting metals such as iron and copper, acting as receptors for hormones, providing structure and support to cells, and participating in immune response as antibodies. From these few examples of their numerous functions, it is easy to see that proteins are truly the physical basis of life.24
Basic Structure
Proteins consist of the elements carbon, oxygen, hydrogen, nitrogen, and sulfur. It is the fact that proteins contain nitrogen that sets them apart from pure carbohydrates and lipids, which do not contain nitrogen atoms. Proteins are polymers built from one or more unbranched chains of amino acids. A typical protein contains 200 to 300 amino acids, but some are much smaller (peptides) and some are much larger (e.g., titin, in muscle). Proteins are considered macromolecules and range in molecular mass from approximately 6,000 Daltons for insulin to several million Daltons for some structural proteins.
There are four distinct levels of a protein’s structure: primary, secondary, tertiary, and quaternary. Primary structure represents the number and types of amino acids in the specific amino acid sequence. In order to function properly, proteins must have the correct sequence of amino acids. For example, when the amino acid valine is substituted for glutamic acid in the α-chain of hemoglobin A, hemoglobin S is formed, which results in sickle cell anemia.
Secondary structure refers to commonly formed structures stabilized by hydrogen bonds between the amino acids within the protein. Common secondary structures are the α-helix, β-pleated sheet, and turns, with most serum proteins forming a helix.25 Secondary structures add new properties to a protein such as strength and flexibility (Fig. 11.4).
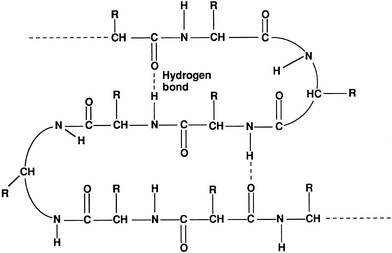
FIGURE 11.4 Secondary structure of proteins.
Tertiary structure refers to the overall shape, or conformation, of the protein molecule. The conformation is known as the fold, or the spatial relationship of the secondary structures to one another. Tertiary structures are three dimensional and result from the interaction of side chains, which are stabilized through the hydrophobic effect, ionic attraction, hydrogen bonds, and disulfide bonds. The function and physical and chemical properties of a protein are related to its tertiary structure.26
Quaternary structure is the shape or structure that results from the interaction of more than one protein molecule, or protein subunits, held together by noncovalent forces such as hydrogen bonds and electrostatic interactions.
General Chemical Properties
The structure of a protein directly affects its chemical properties. Proteins contain many ionizable groups on the side chains of their amino acids as well as on their N- and C-terminal ends. As a result, proteins can be positively and negatively charged. The side chains of lysine, arginine, and histidine include basic groups, and acidic groups are found on the side chains of glutamate, aspartic acid, cysteine, and tyrosine. The acid or base groups that are not involved in the peptide bond can exist in different charged forms depending on the pH of the surrounding environment (Fig. 11.5). The pH of the solution, the pKa of the side chain, and the side chain’s environment influence the charge on each side chain. The relationship between pH, pKa, and charge for individual amino acids can be described by the Henderson-Hasselbalch equation:
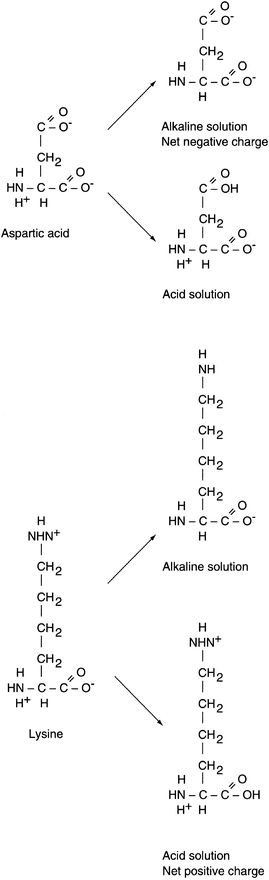
FIGURE 11.5 Charged states of amino acids.
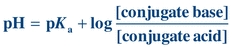 (Eq. 11-1)
(Eq. 11-1) In general terms, as the pH of a solution increases, becoming more alkaline, deprotonation of the acidic and basic groups occurs; carboxyl groups are converted to carboxylate anions (R–COOH to R–COO–) and ammonium groups are converted to amino groups (R–NH3+ to R–NH2). The pH at which an amino acid or protein has no net charge is known as its isoelectric point (pI). When the pH is greater than the pI, the protein has a net negative charge, and when the pH is less than the pI, the protein has a net positive charge. Therefore, the pI is the point at which the number of positively charged groups equals the number of negatively charged groups in a protein. If a protein is placed in a solution that has a pH greater than the pI, the protein will be negatively charged. If the protein is placed in a solution that has a pH less than the pI, the protein will be positively charged. Proteins differ in their pI values, but for most proteins it occurs in the pH range of 5.5 to 8. Because proteins carry different net charges depending on the pH, the difference in net charge at a given pH is the basis for several procedures used to separate and quantify proteins.
The net charge on the surface of a protein depends on the number and type of amino acids it contains as well as the pH of its environment. A protein’s solubility is dependent on the charge on its surface with its lowest solubility at its pI where the positive and negative charges balance and the net charge is zero. If there is a charge at the protein surface, the protein is referred to as hydrophilic and prefers to interact with water rather than with other protein molecules making it more soluble. The solubility of proteins in blood requires a pH in the range of 7.35 to 7.45. Without a net surface charge, the protein is less soluble and protein–protein interactions and precipitation are more likely. Some laboratory methods rely on the solubility of proteins to separate and quantify them.
When the secondary, tertiary, or quaternary structure of a protein is disturbed, the protein may lose its functional and chemical characteristics. This loss of its native, or naturally occurring, folded structure is called denaturation. Denaturation can be caused by heat, hydrolysis by strong acid or alkali, enzymatic action, exposure to urea or other substances, or exposure to ultraviolet light.
Synthesis
Most plasma proteins are synthesized in the liver and secreted by the hepatocyte into circulation. Immunoglobulins are an exception because they are synthesized in plasma cells. It is the information encoded in genes that provides each protein with its own unique amino acid sequence. The amino acid sequence of a polypeptide chain is determined by a corresponding sequence of bases (guanine, cytosine, adenine, and thymine) in the DNA contained in the specific gene. This genetic code is a set of three nucleotides known as codons, with each three-nucleotide combination standing for a specific amino acid. Because DNA contains four nucleotides, the total number of possible codons is 64; therefore, some redundancy in the genetic code allows for some amino acids to be specified by more than one codon. Double-stranded DNA unfolds in the nucleus, and one strand is used as a template for the formation of a complementary strand of mRNA in a process known as transcription. The mRNA is manufactured in the cell nucleus and then translocated across the nuclear membrane into the cytoplasm for translation.
In the process of translation, the mRNA strand is used as a template for protein synthesis by the ribosome. The mRNA is loaded onto the ribosome and is read three nucleotides at a time by matching each codon to its base pairing anticodon located on a tRNA molecule. tRNA is a short chain of RNA that occurs freely in the cytoplasm. Each amino acid has a specific tRNA that contains three bases corresponding to the three bases in the mRNA. The tRNA carries its particular amino acid to the ribosome and attaches to the mRNA in accordance with the matching codon. As each new tRNA brings in the next amino acid, the preceding amino acid is transferred onto the amino group of the new amino acid and enzymes located in the ribosomes form a peptide bond. The tRNA is released into the cytoplasm, where it can pick up another amino acid, and the cycle repeats. In this manner, the amino acids are aligned in sequence and joined together. This process continues until the mRNA message is read and all amino acids are in the specific sequence to form the polypeptide chain. When the terminal codon is reached, the peptide chain is detached and the ribosome and mRNA dissociate. Figure 11.6 illustrates basic protein synthesis.
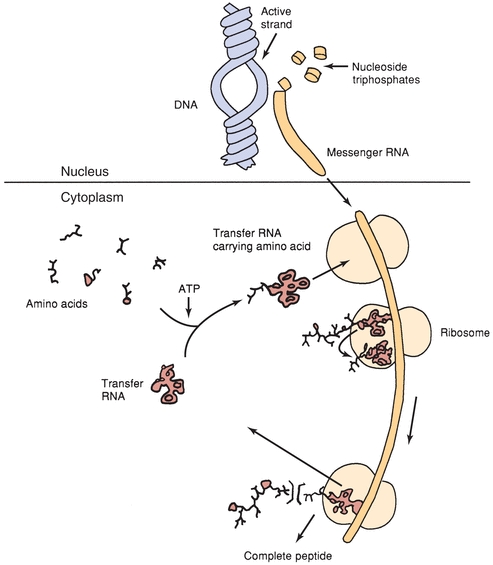
FIGURE 11.6 Schematic summary of protein synthesis.
Protein synthesis occurs at a rate of approximately two to six peptide bonds per second. Intracellular proteins are generally synthesized on free ribosomes, whereas proteins made by the liver for secretion are made on ribosomes attached to the rough endoplasmic reticulum. In some tissues, hormones, such as thyroxine, growth hormone, insulin, and testosterone, may assist in controlling protein synthesis.
Catabolism and Nitrogen Balance
Unlike fats and carbohydrates, nitrogen has no designated storage, and dietary proteins play an essential role in providing all the essential amino acids. Insufficient dietary quantities of even one amino acid can quickly limit synthesis of many essential proteins.27 Most proteins in the body are repetitively synthesized and then degraded allowing for efficient recycling of amino acids. A balance exists between protein synthesis (anabolism) and protein breakdown (catabolism) and the turnover totals about 125 to 220 g of protein each day, with the rate of individual proteins varying widely. For example, plasma proteins and most intracellular proteins are rapidly degraded, having half-lives of hours or days, whereas some of the structural proteins, such as collagen, are metabolically stable and have half-lives of years. In health, nitrogen balance is maintained by equal intake and excretion of amino acids. Pregnant women, growing children, and adults recovering from major illness are often in positive nitrogen balance, which means that their nitrogen intake exceeds their loss as net protein synthesis proceeds. When more nitrogen is excreted than is incorporated into the body, an individual is in negative nitrogen balance. A negative nitrogen balance may occur in conditions in which there is excessive tissue destruction, such as burns, wasting diseases, continual high fevers, or starvation.
The breakdown of protein occurs in the digestive tract and kidneys, but primarily in the liver. Nitrogen elimination begins intracellularly with protein degradation. There are two main routes for converting intracellular proteins to free amino acids: a lysosomal pathway and a cytosolic pathway.
The central reactions that remove amino acid nitrogen from the body are known as transaminations. Transaminations involve moving an α-amino group from a donor α-amino acid to the keto carbon of an acceptor α-ketoacid. These reversible reactions are catalyzed by a group of intracellular enzymes known as transaminases and result in the production of ammonia and ketoacids. The ammonia is converted to urea by the urea cycle in hepatocytes, which is excreted in the urine. The ketoacids are oxidized by means of the citric acid cycle and are converted into glucose or fat.
Classification
From 2003 to 2005, the Human Plasma Proteome Project directed by the Human Plasma Proteome Organization (HUPO) prepared and distributed reference specimens of human serum and plasma to 55 participating laboratories worldwide. Some of the long-term goals of this project are the comprehensive analysis of plasma and serum protein constituents in humans and their physiologic, pathologic, and pharmacologic applications. Protocols used combinations of depletion, fractionation, mass spectrometry, and immunoassay methods linked via search engines and annotation groups to gene and protein databases. A new human plasma proteome database was created with, obviously, much work yet to be done.28 The findings from the collaborative project and from laboratory-specific ancillary projects were published in a special issue of Proteomics, “Exploring the Human Plasma Proteome” in August 2005.
Generally speaking, proteins do everything in the living cell.29 Proteins are responsible for many different functions within cells so they are commonly classified by the function(s) they perform.
Enzymes
Enzymes are proteins that catalyze biochemical reactions. They are normally found intracellularly, but are released into the bloodstream as a result of tissue damage, making enzyme measurements an important diagnostic tool. Transaminases, dehydrogenases, and phosphatases are just a few examples of enzyme groups that are routinely tested in the clinical laboratory to evaluate possible tissue damage.
Hormones
Hormones are chemical messenger proteins that control the action(s) of specific cells or organs. Hormones directly affect growth and development, metabolism, sexual function, reproduction, and behavior. Examples of hormones that are commonly tested in the clinical laboratory in blood, urine, or saliva are insulin, testosterone, growth hormone, follicle-stimulating hormone, and cortisol.
Transport Proteins
Many proteins are involved in the transport of ions, small molecules, or macromolecules, such as hormones, vitamins, minerals, and lipids, across a biologic membrane. Examples of commonly measured transport proteins are hemoglobin, albumin, ceruloplasmin, haptoglobin, and transferrin.
Immunoglobulins
Immunoglobulins or antibodies are proteins that are produced by B cells (lymphocytes) in the bone marrow. Immunoglobulins mediate the humoral immune response to identify and neutralize foreign antigens. Examples of immunoglobulins of clinical importance are IgG, IgM, IgE, and IgA.
Structural Proteins
Fibrous proteins provide structure to many cells and tissues throughout the body, such as muscle, tendons, and bone matrix. Collagen, elastin, and keratin are examples of structural proteins.
Storage Proteins
Storage proteins serve as reservoirs for metal ions and amino acids so they can be stored without causing harm and released later. Ferritin is a commonly measured protein that stores iron for later use in the manufacture of hemoglobin.
Energy Source
Some proteins serve as an energy source for tissues and muscle. Creatine is one example of an energy source protein as it helps to supply energy to cells throughout the body, but is primarily found in muscle tissue.
Osmotic Force
Some proteins function in the distribution of water throughout the compartments of the body. Their colloid osmotic force, due to their size, does not allow proteins to cross the capillary membranes. As a result, water is absorbed from the tissue into the venous portion of the capillary. When the concentration of plasma proteins is significantly decreased, the concomitant decrease in the plasma colloidal osmotic (oncotic) pressure results in increased levels of interstitial fluid and edema. This often occurs in renal disease when proteins are inappropriately excreted in urine and plasma protein concentrations are decreased.
Protein functions are summarized in Table 11.2.
TABLE 11.2 Functions of Proteins

Proteins may also be classified according to their structure. Simple proteins contain peptide chains composed of only amino acids. Simple proteins may be globular or fibrous in shape. Globular proteins are globe-like and have symmetrical proteins that are soluble in water. Globular proteins function as transporters, enzymes, and messengers. Examples of globular proteins are albumin, hemoglobin, and the immunoglobulins, IgG, IgA, and IgM. Fibrous proteins form long protein filaments or subunits, are asymmetrical and usually inert, and are generally water insoluble due to their hydrophobic R groups. Fibrous proteins provide structure to cells, such as connective tissues, tendons, bone, and muscle. Examples of fibrous proteins include troponin and collagen. Conjugated proteins consist of a protein and a nonprotein. The nonamino part of a conjugated protein is generally referred to as the prosthetic group. The prosthetic group may be a lipid, carbohydrate, porphyrin, metal, etc. It is the prosthetic group that defines the characteristics of a conjugated protein. Examples of conjugated proteins are metalloproteins, glycoproteins, lipoproteins, and nucleoproteins. Metalloproteins have a metal ion attached to the protein, either directly, as in ferritin, which contains iron, and ceruloplasmin, which contains copper, or as a complex metal such as hemoglobin and flavoproteins. Lipoproteins are composed of a protein and a lipid such as cholesterol or triglyceride; examples of lipoproteins include high-density lipoproteins (HDLs) and low-density lipoproteins (LDLs). There are several terms used to describe conjugated proteins that are bound to carbohydrates. Generally, molecules with a composition of 10% to 40% carbohydrate are referred to as glycoproteins. Examples of glycoproteins are haptoglobin and α1-antitrypsin. When the percentage of carbohydrate is greater than 40%, the protein conjugate is referred to as a mucoprotein or proteoglycan. An example of a mucoprotein is mucin, which is a lubricant that protects body surfaces from friction or erosion. Increased mucin production occurs in many adenocarcinomas, including cancer of the pancreas, lung, breast, ovary, and colon. Moreover, mucins are also being investigated for their potential as diagnostic markers.30 Nucleoproteins are those proteins that are combined with nucleic acids. Chromatin is an example of a nucleoprotein as it is a complex of DNA and protein that makes up chromosomes.
Several databases have been developed to aid in the identification of proteins based on their structure. The Structural Classification of Proteins (SCOP) database was created to provide a detailed and comprehensive description of the structural domains of proteins based on similarities in their amino acid sequences and three-dimensional structures. SCOP utilizes four levels of structural classification: class, fold, superfamily, and family. Originally published in 1995, SCOP is usually updated at least annually by Alexei G. Murzin et al., upon whose expertise the classification rests.31 The Families of Structurally Similar Proteins (FSSP), also known as Fold classification, is based on the structure–structure alignment of proteins and a three-dimensional comparison of protein structures in the Protein Data Bank. Alignments and classification are updated continuously by the Dali (Distance matrix ALIgnment) server, which is an automatic server that makes a three-dimensional comparison of protein structures.32
PLASMA PROTEINS
Plasma proteins are the most commonly analyzed proteins in the clinical laboratory and can be divided into two major groups: albumin and globulins. There are four major types of globulins with specific properties and functions. Analysis of blood specimens will typically include four protein measurements: total protein, albumin, globulins, and the albumin-to-globulin (A/G) ratio; however, there are many other clinically important proteins that may be measured when indicated by clinical presentation. Some of the more significant plasma proteins and their function, structure, and relation to disease states are discussed below. The characteristics of select plasma proteins are listed in Table 11.3.
TABLE 11.3 Characteristics of Selected Plasma Proteins
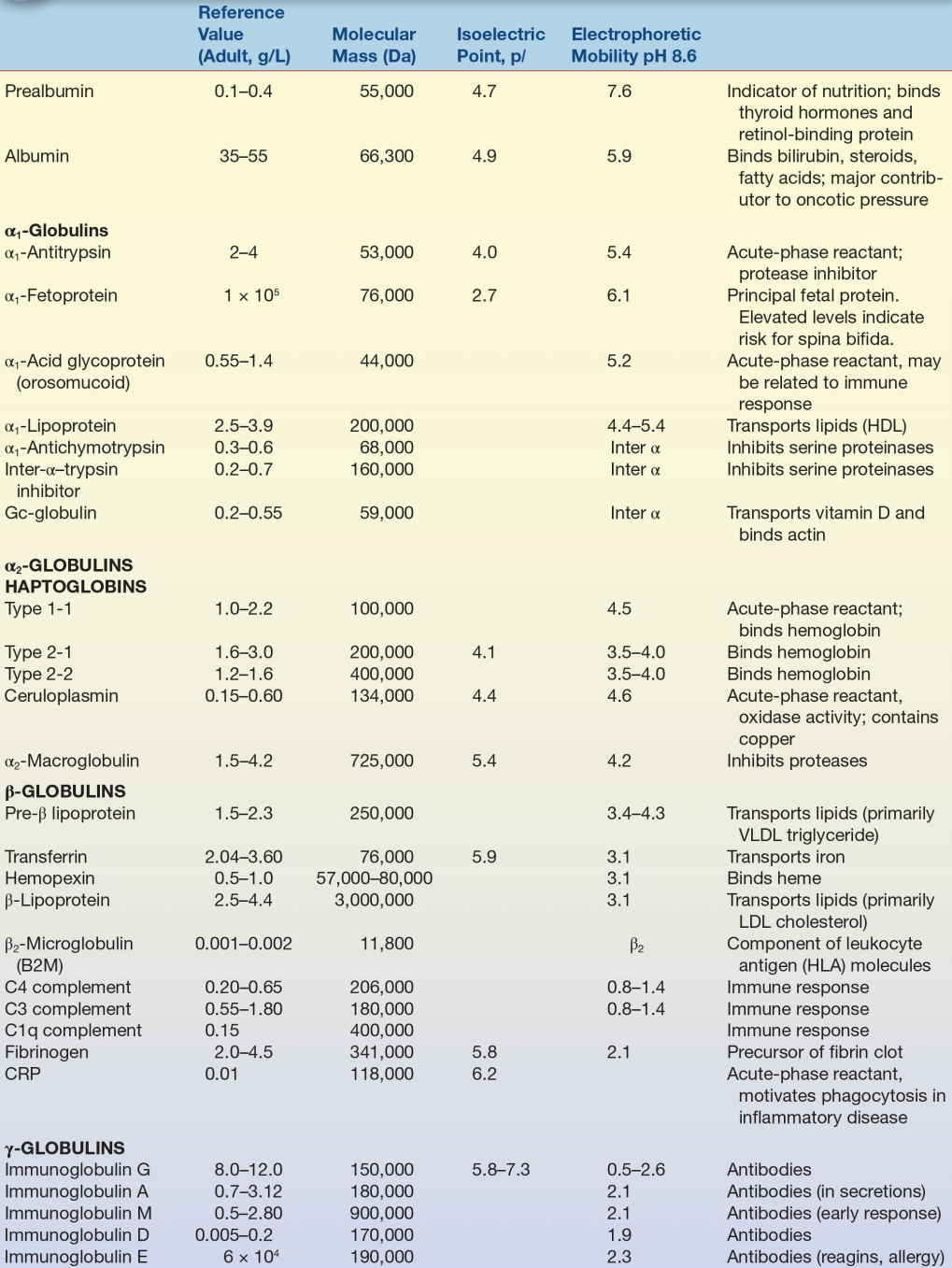
HDL, high-density lipoproteins; VLDL, very-low-density lipoprotein; HLA, human leukocyte antigen; CRP, C-reactive protein.
Prealbumin
Prealbumin, or transthyretin, is so named because it migrates before albumin in classic serum protein electrophoresis (SPE). It can also be separated using high-resolution electrophoresis (HRE) or immunoelectrophoresis techniques. Prealbumin is a transport protein for the thyroid hormones, thyroxine (T4), and triiodothyronine (T3). Prealbumin also forms a complex with retinol-binding protein to transport retinol (vitamin A) and is rich in the amino acid tryptophan. Serum and plasma prealbumin concentrations may be decreased in hepatic damage due to decreased protein synthesis, during an acute-phase inflammatory response, or as a result of tissue necrosis. A low prealbumin may also indicate poor nutritional status. Diets deficient in protein may not provide sufficient amino acids for adequate protein synthesis by the liver resulting in decreased plasma concentrations of prealbumin, albumin, and β-globulins. Prealbumin has a half-life of approximately 2 days, so blood concentrations of prealbumin decrease faster than do those of other proteins with a longer half-life when protein synthesis is inhibited. Blood concentrations of prealbumin may be increased in patients receiving steroid therapy, who have issues with alcohol abuse, or who are in chronic renal failure.
Albumin
Albumin is synthesized in the liver at a rate of 9 to 12 g/day and is the most abundant protein in the plasma. Albumin also exists in the extravascular (interstitial) space. The total amount of extravascular albumin exceeds the total intravascular amount by about 30%; however, the concentration of albumin in plasma (albumin mass/plasma volume) is much greater than its concentration in the interstitial space. Albumin leaves the bloodstream at a rate of 4% to 5% of the total intravascular albumin concentration per hour. This rate of movement is known as the transcapillary escape rate, which measures systemic capillary efflux of albumin. Albumin is responsible for nearly 80% of the colloid osmotic pressure (COP) of the intravascular fluid, which maintains the appropriate fluid balance in the tissue. Albumin also buffers pH and is a negative acute-phase reactant protein.
A primary function of albumin is its capacity to bind and transport various substances in the blood. There are four binding sites on albumin that have varying specificities for different substances. Albumin is involved in the transport of thyroid hormones, unconjugated bilirubin, fat-soluble hormones, iron, fatty acids, calcium (Ca2+), magnesium (Mg2+), and many drugs such as salicylic acid (aspirin). Albumin also binds certain dyes, which is the basis for many of the methods used for quantitation of albumin.
Several recent studies have focused on the clinical applications of glycated (or glycosylated) albumin as a more sensitive indicator of short-term hyperglycemic control than glycated hemoglobin. Glycated hemoglobin represents trends in blood glucose over a period of 3 months or approximately 120 days, which is the life span of red blood cells. As the half-life of serum albumin is 20 days, glycated albumin represents serum glucose patterns approximating 1 month. Affinity chromatographic methods based on specific interaction of boronic acids with glycated proteins have been applied to determine serum concentrations of glycated albumin.33
Decreased blood concentrations of albumin are most commonly associated with an acute inflammatory response as albumin is a negative acute-phase reactant. Liver and kidney disease may also result in low blood albumin concentrations. The liver is a primary site for protein synthesis, and damage to hepatocytes, such as in liver cirrhosis, may result in decreased protein synthesis. Note that an increase in globulins occurs in early liver cirrhosis, which balances the loss in albumin to give a total protein concentration within acceptable limits. Albumin is normally excreted in very small amounts by the kidneys; however, increased renal loss of albumin commonly occurs in renal disease. This increased excretion occurs when the glomerulus no longer restricts the passage of proteins from the blood into the ultrafiltrate as occurs in nephrotic syndrome. Low albumin concentrations may also be the result of malnutrition and malabsorption in which an inadequate ingestion of proteins or amino acid-rich foods results in decreased protein synthesis by the liver. Less commonly, low blood albumin concentrations occur as a result of hypothyroidism, burns or exfoliative dermatitis, dilution by excessive intake (polydipsia), or infusion of liquids (intravenously). Albumin may be redistributed by hemodilution, increased capillary permeability, or decreased lymph clearance. In sepsis, there is a profound reduction in plasma albumin associated with marked fluid shifts. Mutations resulting from an autosomal recessive trait can cause an absence of albumin, known as analbuminemia, or presence of albumin that has unusual molecular characteristics, which is referred to as bisalbuminemia. Bisalbuminemia is demonstrated by the presence of two albumin bands during electrophoresis instead of the single band usually observed. Both analbuminemia and bisalbuminemia are rare.
Abnormally high albumin concentrations are seldom clinically important and are generally associated with dehydration or excessive albumin infusion.34
Globulins
The globulin group of proteins consists of α1, α2, β, and γ fractions. Each fraction consists of a number of different proteins with individual functions. Select examples of globulins from each of the fractions are discussed below.
α1-Antitrypsin
α1-Antitrypsin is a glycoprotein synthesized mainly in the liver. Its main function is in the inhibition of the protease, neutrophil elastase. Neutrophil elastase is released from leukocytes during an infection, but can also destroy alveoli leading to emphysema if not inhibited by α1-antitrypsin. Mutations in the SERPINA1 gene can lead to a deficiency of α1-antitrypsin protein or an abnormal form of the protein that does not properly control neutrophil elastase. The abnormal form of α1-antitrypsin can also accumulate in the liver causing cirrhosis. α1-Antitrypsin is a positive acute-phase reactant; therefore, increased levels of α1-antitrypsin are seen in inflammatory reactions as well as in pregnancy and contraceptive use. Several phenotypes of α1-antitrypsin deficiency have been identified. The most common phenotype is MM (allele PiM) and is associated with normal α1-antitrypsin activity. Other alleles are PiS, PiZ, PiF, and Pi (null). The homozygous phenotype ZZ individual is at greatest risk for developing hepatic and pulmonary disease from α1-antitrypsin deficiency. Replacement therapy using purified α1-antitrypsin protein from pooled plasma can dramatically slow disease progression. It is also recommended that individuals with α1-antitrypsin deficiency do not smoke.
Decreased α1-antitrypsin levels are most often identified in the clinical laboratory by the lack of an α1-globulin band on serum protein electrophoresis because α1-antitrypsin accounts for approximately 90% of the α1-globulin fraction, which migrates immediately following albumin. Quantitative methods used to confirm α1-antitrypsin deficiency are radial immunodiffusion and automated immunonephelometric assays. Phenotyping may also be performed using immunofixation techniques.35
α1-Fetoprotein
α1-Fetoprotein (AFP) is synthesized in utero by the developing embryo and fetus and then by the parenchymal cells of the liver. AFP concentrations decrease gradually after birth, reaching adult concentrations by 8 to 12 months of age. The physiologic function of AFP in adults has not been completely identified, but it has been proposed that α1-fetoprotein protects the developing fetus from immunologic attack by the mother. Conditions associated with an elevated AFP concentration include spina bifida, neural tube defects, abdominal wall defects, anencephaly, general fetal distress, and the presence of twins. Low levels of maternal AFP indicate an increased risk for trisomy 21 (Down syndrome) and trisomy 18 (Edwards syndrome).
AFP screening is performed between 15 and 20 weeks gestation when maternal AFP increases gradually; therefore, interpretation requires accurate dating of the pregnancy. Measurements of AFP can be affected by laboratory-specific technique, which can result in difficulty comparing absolute results between centers. AFP levels are also affected by maternal weight, race, and diabetes; therefore, test results need to be adjusted for these variables. To account for these maternal variables, the multiple of the median or MoM is utilized. MoM is a reflection of an individual patient’s value compared with the median. MoM is calculated by dividing the patient’s AFP value by the median reference value for that gestational age. Most screening laboratories use 2.0 MoM as the upper limit and 0.5 MoM as the lower limit of normal for maternal serum. The methods commonly used for AFP determinations are radioimmunoassay (RIA) and enzyme-labeled immunoassay (EIA). Maternal screening tests have been established as a triple or quadruple test group using a mathematical calculation involving the concentrations of AFP, human chorionic gonadotropin (hCG), unconjugated estriol, and inhibin A. These calculations are used to determine a numerical risk for chromosomal abnormalities in the fetus, which can be compared with an established cutoff. The interpretation of these test results should be provided by a genetic counselor or clinician to aid parents and their physicians in making decisions about the management of the pregnancy.
AFP may also be used as a tumor marker and is fractionated by affinity electrophoresis into three isoforms (L1, L2, and L3) based on their reactivity with the lectin Lens culinaris agglutinin (LCA). AFP-L3 is now being considered as a tumor marker for the North American population for screening chronic liver disease patients for hepatocellular carcinoma (HCC). Results for AFP-L3 are reported as a ratio of LCA-reactive AFP to total AFP (AFP-L3%). Studies have shown that AFP-L3% test results of greater than 10% are associated with a sevenfold increase in the risk of developing HCC within the next 21 months.36 AFP’s utility as a tumor marker will be discussed further in Chapter 32.
α1-Acid Glycoprotein
α1-Acid glycoprotein (AAG), or orosomucoid, is a major plasma glycoprotein that is negatively charged even in acidic solutions lending to its name. This protein is produced by the liver and is a positive acute-phase reactant. The possibility that AAG regulates immune responses has been suggested by several findings.37 There is a strong similarity in the amino acid sequences between AAG and immunoglobulin. AAG elevates following stress, inflammation, tissue damage, acute myocardial infarction (AMI), trauma, pregnancy, cancer, pneumonia, rheumatoid arthritis, and surgery. Serum AAG concentrations are also used in diagnosis and evaluation of neonatal bacterial infections. In the past decade, the binding of drugs to this plasma protein has become increasingly important in regard to drug action, distribution, and disposition. The analytic methods used most commonly for the determination of AAG are radial immunodiffusion, immunoturbidity, and nephelometry. Immunofixation techniques have been used to study inherited variants. The reference interval for healthy individuals is 50 to 120 mg/dL.
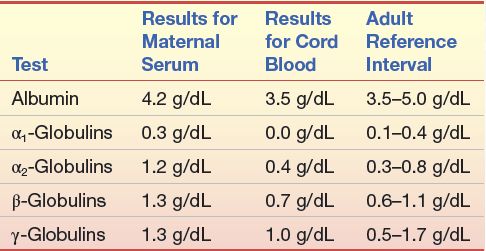
CASESTUDY 11 .1
Immediately following the birth of a baby girl, the attending physician requested a protein electrophoretic examination of the mother’s serum. This was done on a sample that was obtained on the mother’s admission to the hospital the previous day. An electrophoretic examination was also performed on the cord blood specimen. Laboratory reports are shown in Case Study Table 11.1.1.
The appearance of the mother’s electrophoretic pattern was within that expected for a healthy person. The electrophoretic pattern of the cord blood serum resembled the one shown in Figure 11.8C.
CASE STUDY TABLE 11.1.1 Electrophoresis Values
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


