(Eq. 6-1)
Modern partition chromatography uses pseudoliquid stationary phases that are chemically bonded to the support or high-molecular-weight polymers that are insoluble in the mobile phase.1 Partition systems are considered normal phase when the mobile solvent is less polar than the stationary solvent and reverse phase when the mobile solvent is more polar.
Partition chromatography is applicable to any substance that may be distributed between two liquid phases. Because ionic compounds are generally soluble only in water, partition chromatography works best with nonionic compounds.
Steric Exclusion
Steric exclusion, a variation of liquid–solid chromatography, is used to separate solute molecules on the basis of size and shape. The chromatographic column is packed with porous material, as shown in Figure 6.1. A sample containing different-sized molecules moves down the column dissolved in the mobile solvent. Small molecules enter the pores in the packing and are momentarily trapped. Large molecules are excluded from the small pores and so move quickly between the particles. Intermediate-sized molecules are partially restricted from entering the pores and, therefore, move through the column at an intermediate rate that is between those of the large and small molecules.
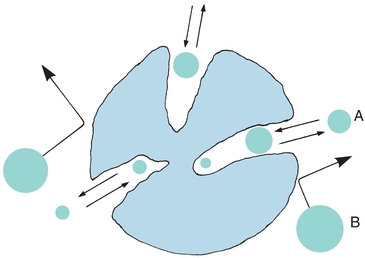
FIGURE 6.1 Pictorial concept of steric exclusion chromatography. Separation of sample components by their ability to permeate pore structure of column-packing material. Smaller molecules (A) permeating the interstitial pores; large excluded molecules (B). (Adapted from Parris NA. Instrumental Liquid Chromatography: A Practical Manual on High Performance Liquid Chromatographic Methods. New York, NY: Elsevier; 1976.)
Early methods used hydrophilic beads of cross-linked dextran, polyacrylamide, or agarose, which formed a gel when soaked in water. This method was termed gel filtration. A similar separation process using hydrophobic gel beads of polystyrene with a nonaqueous mobile phase was called gel permeation chromatography. Current porous packing uses rigid inorganic materials such as silica or glass. The term steric exclusion includes all these variations. Pore size is controlled by the manufacturer, and packing materials can be purchased with different pore sizes, depending on the size of the molecules being separated.
Ion-Exchange Chromatography
In ion-exchange chromatography, solute mixtures are separated by virtue of the magnitude and charge of ionic species. The stationary phase is a resin, consisting of large polymers of substituted benzene, silicates, or cellulose derivatives, with charged functional groups. The resin is insoluble in water, and the functional groups are immobilized as side chains on resin beads that are used to fill the chromatographic column. Figure 6.2A shows a resin with sulfonate functional groups. Hydrogen+ ions are loosely held and free to react. This is an example of a cation-exchange resin. When a cation such as Na+ comes in contact with these functional groups, an equilibrium is formed, following the law of mass action. Because there are many sulfonate groups, Na+ is effectively and completely removed from solution. The Na+ concentrated on the resin column can be eluted from the resin by pouring acid through the column, driving the equilibrium to the left.
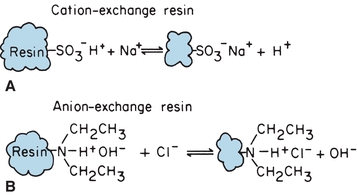
FIGURE 6.2 Chemical equilibrium of ion-exchange resins. (A) Cation-exchange resin. (B) Anion-exchange resin.
Anion-exchange resins are made with exchangeable hydroxyl ions such as the diethylamine functional group illustrated in Figure 6.2B. They are used like cation-exchange resins, except that hydroxyl ions are exchanged for anions. The example shows Cl− in sample solution exchanged for OH− from the resin functional group. Anion and cation resins mixed together (mixed-bed resin) are used to deionize water. The displaced protons and hydroxyl ions combine to form water. Ionic functional groups other than the illustrated examples are used for specific analytic applications. Ion-exchange chromatography is used to remove interfering substances from a solution, to concentrate dilute ion solutions, and to separate mixtures of charged molecules, such as amino acids. Changing pH and ionic concentration of the mobile phase allows separation of mixtures of organic and inorganic ions.
Chromatographic Procedures
Thin-Layer Chromatography
Thin-layer chromatography (TLC) is a variant of column chromatography. A thin layer of sorbent, such as alumina, silica gel, cellulose, or cross-linked dextran, is uniformly coated on a glass or plastic plate. Each sample to be analyzed is applied as a spot near one edge of the plate, as shown in Figure 6.3. The mobile phase (solvent) is usually placed in a closed container until the atmosphere is saturated with solvent vapor. One edge of the plate is placed in the solvent, as shown. The solvent migrates up the thin layer by capillary action, dissolving and carrying sample molecules. Separation can be achieved by any of the four processes previously described, depending on the sorbent (thin layer) and solvent chosen. After the solvent reaches a predetermined height, the plate is removed and dried. Sample components are identified by comparison with standards on the same plate. The distance a component migrates, compared with the distance the solvent front moves, is called the retention factor, Rf:
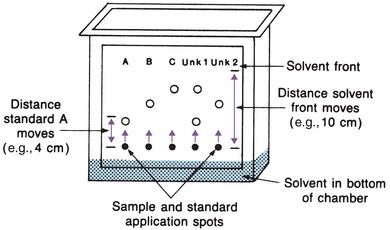
FIGURE 6.3 Thin-layer chromatography plate in chromatographic chamber.
 (Eq. 6-2)
(Eq. 6-2) Each sample component Rf is compared with the Rf of standards. Using Figure 6.3 as an example, standard A has an Rf value of 0.4, standard B has an Rf value of 0.6, and standard C has an Rf value of 0.8. The first unknown contains A and C, because the Rf values are the same. This ratio is valid only for separations run under identical conditions. Because Rf values may overlap for some components, further identifying information is obtained by spraying different stains on the dried plate and comparing colors of the standards.
TLC is most commonly used as a semiquantitative screening test. Technique refinement has resulted in the development of semiautomated equipment and the ability to quantitate separated compounds. For example, sample applicators apply precise amounts of sample extracts in concise areas. Plates prepared with uniform sorbent thickness, finer particles, and new solvent systems have resulted in the technique of high-performance thin-layer chromatography (HPTLC).2 Absorbance of each developed spot is measured using a densitometer, and the concentration is calculated by comparison with a reference standard chromatographed under identical conditions.
High-Performance Liquid Chromatography
Modern high-performance liquid chromatography (HPLC) uses pressure for fast separations, controlled temperature, in-line detectors, and gradient elution techniques.3,4 Figure 6.4 illustrates the basic components.
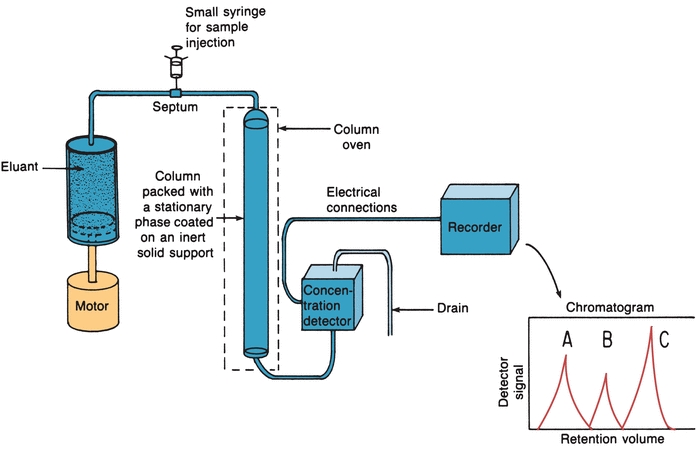
FIGURE 6.4 High-performance liquid chromatography basic components. (Adapted from Bender GT. Chemical Instrumentation: A Laboratory Manual Based on Clinical Chemistry. Philadelphia, PA: WB Saunders; 1972.)
Pumps
A pump forces the mobile phase through the column at a much greater velocity than that accomplished by gravity flow columns and includes pneumatic, syringe, reciprocating, or hydraulic amplifier pumps. The most widely used pump today is the mechanical reciprocating pump, which is used as a multihead pump with two or more reciprocating pistons. During pumping, the pistons operate out of phase (180° for two heads, 120° for three heads) to provide constant flow. Pneumatic pumps are used for preoperative purposes; hydraulic amplifier pumps are no longer commonly used.
Columns
The stationary phase is packed into long stainless steel columns. HPLC is usually run at ambient temperatures, although columns can be put in an oven and heated to enhance the rate of partition. Fine, uniform column packing results in much less band broadening but requires pressure to force the mobile phase through. The packing can also be pellicular (an inert core with a porous layer), inert and small particles, or macroporous particles. The most common material used for column packing is silica gel. It is very stable and can be used in different ways. It can be used as solid packing in liquid–solid chromatography or coated with a solvent, which serves as the stationary phase (liquid–liquid). As a result of the short lifetime of coated particles, molecules of the mobile-phase liquid are now bonded to the surface of silica particles.
Reversed-phase HPLC is now popular; the stationary phase is nonpolar molecules (e.g., octadecyl C-18 hydrocarbon) bonded to silica gel particles. For this type of column packing, the mobile phase commonly used is acetonitrile, methanol, water, or any combination of solvents. A reversed-phase column can be used to separate ionic, nonionic, and ionizable samples. A buffer is used to produce the desired ionic characteristics and pH for separation of the analyte. Column packings vary in size (3 to 20 mm), using smaller particles mostly for analytic separations and larger ones for preparative separations.
Sample Injectors
A small syringe can be used to introduce the sample into the path of the mobile phase that carries it into the column (Fig. 6.4). The best and most widely used method, however, is the loop injector. The sample is introduced into a fixed-volume loop. When the loop is switched, the sample is placed in the path of the flowing mobile phase and flushed onto the column. Loop injectors have high reproducibility and are used at high pressures. Many HPLC instruments have loop injectors that can be programmed for automatic injection of samples. When the sample size is less than the volume of the loop, the syringe containing the sample is often filled with the mobile phase to the volume of the loop before filling the loop. This prevents the possibility of air being forced through the column because such a practice may reduce the lifetime of the column-packing material.
Detectors
Modern HPLC detectors monitor the eluate as it leaves the column and, ideally, produce an electronic signal proportional to the concentration of each separated component. Spectrophotometers that detect absorbances of visible or UV light are most commonly used. Photodiode array (PDA) and other rapid scanning detectors are also used for spectral comparisons and compound identification and purity. These detectors have been used for drug analyses in urine. Obtaining a UV scan of a compound as it elutes from a column can provide important information as to its identity. Unknowns can be compared against library spectra in a similar manner to MS. Unlike gas chromatography/MS, which requires volatilization of targeted compounds, liquid chromatography (LC)/PDA enables direct injection of aqueous urine samples.
Because many biologic substances fluoresce strongly, fluorescence detectors are also used, involving the same principles discussed in the section on spectrophotometric measurements. Another common HPLC detector is the amperometric or electrochemical detector, which measures current produced when the analyte of interest is either oxidized or reduced at some fixed potential set between a pair of electrodes.
An MS can also be used as a detector, as described later.
Recorders
The recorder is used to record detector signal versus the time the mobile phase passed through the instrument, starting from the time of sample injection. The graph is called a chromatogram (Fig. 6.5). The retention time is used to identify compounds when compared with standard retention times run under identical conditions. Peak area is proportional to concentration of the compounds that produced the peaks.
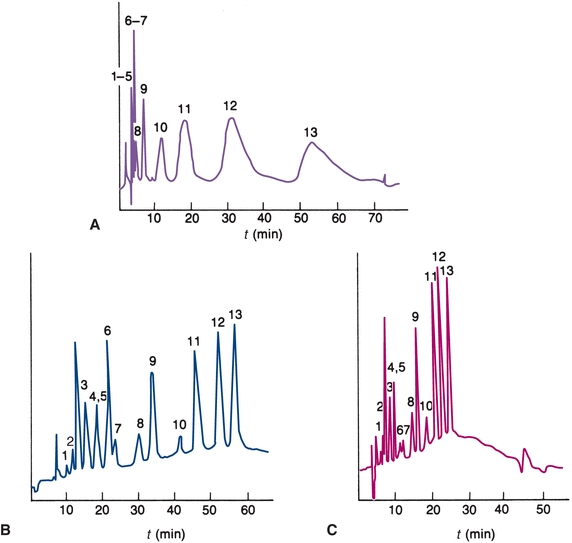
FIGURE 6.5 Chromatograms. (A) Isocratic ion-exchange separation mobile phase contains 0.055 mol/L NaNO3. (B) Gradient elution mobile phase gradient from 0.01 to 0.1 mol/L NaNO3 at 2% per minute. (C)Gradient elution—5% per minute. (Adapted from Horváth C. High Performance Liquid Chromatography, Advances and Perspectives. New York, NY: Academic Press; 1980.)
When the elution strength of the mobile phase is constant throughout the separation, it is called isocratic elution. For samples containing compounds of widely differing relative compositions, the choice of solvent is a compromise. Early eluting compounds may have retention times close to zero, producing a poor separation (resolution), as shown in Figure 6.5A. Basic compounds often have low retention times because C-18 columns cannot tolerate high pH mobile phases. The addition of cation-pairing reagents to the mobile phase (e.g., octane sulfonic acid) can result in better retention of negatively charged compounds onto the column.
The late-eluting compounds may have long retention times, producing broad bands resulting in decreased sensitivity. In some cases, certain components of a sample may have such a great affinity for the stationary phase that they do not elute at all. Gradient elution is an HPLC technique that can be used to overcome this problem. The composition of the mobile phase is varied to provide a continual increase in the solvent strength of the mobile phase entering the column (Fig. 6.5B). The same gradient elution can be performed with a faster change in concentration of the mobile phase (Fig. 6.5C).
Gas Chromatography
Gas chromatography is used to separate mixtures of compounds that are volatile or can be made volatile.5 GC may be gas–solid chromatography, with a solid stationary phase, or gas–liquid chromatography (GLC), with a nonvolatile liquid stationary phase. GLC is commonly used in clinical laboratories. Figure 6.6 illustrates the basic components of a GC system. The setup is similar to HPLC, except that the mobile phase is a gas and samples are partitioned between a gaseous mobile phase and a liquid stationary phase. The carrier gas can be nitrogen, helium, or argon. The selection of a carrier gas is determined by the detector used in the instrument. The instrument can be operated at a constant temperature or programmed to run at different temperatures if a sample has components with different volatilities. This is analogous to gradient elution described for HPLC.
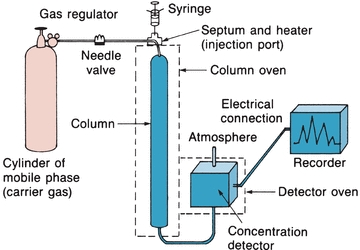
FIGURE 6.6 Gas–liquid chromatography basic components. (Adapted from Bender GT. Chemical Instrumentation: A Laboratory Manual Based on Clinical Chemistry. Philadelphia, PA: WB Saunders; 1972.)
The sample, which is injected through a septum, must be injected as a gas or the temperature of the injection port must be above the boiling point of the components so that they vaporize upon injection. Sample vapor is swept through the column partially as a gas and partially dissolved in the liquid phase. Volatile compounds that are present mainly in the gas phase will have a low partition coefficient and will move quickly through the column. Compounds with higher boiling points will move slowly through the column. The effluent passes through a detector that produces an electric signal proportional to the concentration of the volatile components. As in HPLC, the chromatogram is used both to identify the compounds by the retention time and to determine their concentration by the area under the peak.
Columns
GLC columns are generally made of glass or stainless steel and are available in a variety of coil configurations and sizes. Packed columns are filled with inert particles such as diatomaceous earth or porous polymer or glass beads coated with a nonvolatile liquid (stationary) phase. These columns are usually 1/8 to 1/4 inch wide and 3 to 12 ft long. Capillary wall-coated open tubular columns have inside diameters in the range of 0.25 to 0.50 mm and are up to 60 m long. The liquid layer is coated on the walls of the column. A solid support coated with a liquid stationary phase may in turn be coated on column walls. The liquid stationary phase must be nonvolatile at the temperatures used, must be thermally stable, and must not react chemically with the solutes to be separated. The stationary phase is termed nonselective when separation is primarily based on relative volatility of the compounds. Selective liquid phases are used to separate polar compounds based on relative polarity (as in liquid–liquid chromatography).
Detectors
Although there are many types of detectors, only thermal conductivity (TC) and flame ionization detectors are discussed because they are the most stable (Fig. 6.7). TC detectors contain wires (filaments) that change electrical resistance with change in temperature. The filaments form opposite arms of a Wheatstone bridge and are heated electrically to raise their temperature. Helium, which has a high TC, is usually the carrier gas. Carrier gas from the reference column flows steadily across one filament, cooling it slightly. Carrier gas and separated compounds from the sample column flow across the other filament. The sample components usually have a lower TC, increasing the temperature and resistance of the sample filament. The change in resistance results in an unbalanced bridge circuit. The electrical change is amplified and fed to the recorder. The electrical change is proportional to the concentration of the analyte. Flame ionization detectors are widely used in the clinical laboratory. They are more sensitive than TC detectors. The column effluent is fed into a small hydrogen flame burning in excess air or atmospheric oxygen. The flame jet and a collector electrode around the flame have opposite potentials. As the sample burns, ions form and move to the charged collector. Thus, a current proportional to the concentration of the ions is formed and fed to the recorder.
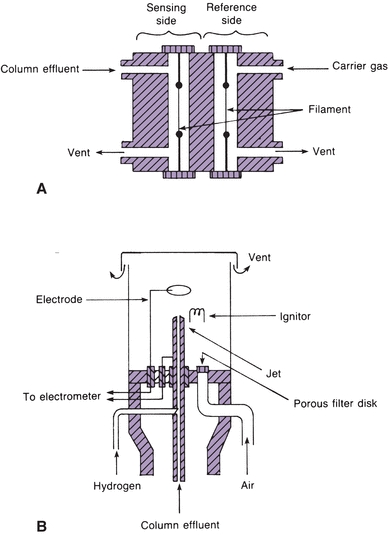
FIGURE 6.7 (A) Schematic diagram of a thermal conductivity detector. (B) Schematic diagram of a flame ionization detector. (Adapted from Tietz NW, ed. Fundamentals of Clinical Chemistry. Philadelphia, PA: WB Saunders; 1987).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


