UNSTABLE HEMOGLOBINS
Amino acid substitutions that reduce solubility or increase susceptibility to oxidation result in unstable hemoglobins that precipitate, forming inclusion bodies injurious to the RBC membrane. Representative mutations are those that interfere with contact points between the α and β subunits (e.g., Hb Philly [β35Tyr→Phe]), alter the helical segments (e.g., Hb Genova [β28Leu→Pro]), or disrupt interactions of the hydrophobic pockets of the globin subunits with heme (e.g., Hb Köln [β98Val→Met]) (Table 127-3). The inclusions, called Heinz bodies, are clinically detectable by staining with supravital dyes such as crystal violet. Removal of these inclusions by the spleen generates pitted, rigid cells that have shortened life spans, producing hemolytic anemia of variable severity, sometimes requiring chronic transfusion support. Splenectomy may be needed to correct the anemia. Leg ulcers and premature gallbladder disease due to bilirubin loading are frequent stigmata.
REPRESENTATIVE ABNORMAL HEMOGLOBINS WITH ALTERED SYNTHESIS OR FUNCTION |
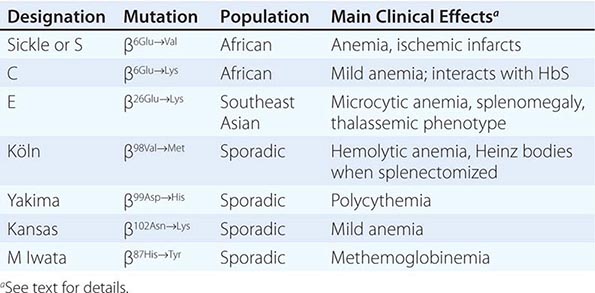
Unstable hemoglobins occur sporadically, often by spontaneous new mutations. Heterozygotes are often symptomatic because a significant Heinz body burden can develop even when the unstable variant accounts for only a portion of the total hemoglobin. Symptomatic unstable hemoglobins tend to be β-globin variants, because sporadic mutations affecting only one of the four α globins alleles would generate only 20–30% abnormal hemoglobin.
HEMOGLOBINS WITH ALTERED OXYGEN AFFINITY
High-affinity hemoglobins (e.g., Hb Yakima [β99Asp→His]) bind oxygen more readily but deliver less O2 to tissues at normal capillary Po2 levels (Fig. 127-2). Mild tissue hypoxia ensues, stimulating RBC production and erythrocytosis (Table 127-3). In extreme cases, the hematocrits can rise to 60–65%, increasing blood viscosity and producing typical symptoms (headache, somnolence, or dizziness). Phlebotomy may be required. Typical mutations alter interactions within the heme pocket or disrupt the Bohr effect or salt-bond site. Mutations that impair the interaction of HbA with 2,3-BPG can increase O2 affinity because 2,3-BPG binding lowers O2 affinity.
Low-affinity hemoglobins (e.g., Hb Kansas [β102Asn→Lys]) bind sufficient oxygen in the lungs, despite their lower oxygen affinity, to achieve nearly full saturation. At capillary oxygen tensions, they lose sufficient amounts of oxygen to maintain homeostasis at a low hematocrit (Fig. 127-2) (pseudoanemia). Capillary hemoglobin desaturation can also be sufficient to produce clinically apparent cyanosis. Despite these findings, patients usually require no specific treatment.
METHEMOGLOBINEMIAS
Methemoglobin is generated by oxidation of the heme iron moieties to the ferric state, causing a characteristic bluish-brown muddy color resembling cyanosis. Methemoglobin has such high oxygen affinity that virtually no oxygen is delivered. Levels >50–60% are often fatal.
Congenital methemoglobinemia arises from globin mutations that stabilize iron in the ferric state (e.g., HbM Iwata [α87His→Tyr], Table 127-3) or from mutations that impair the enzymes that reduce methemoglobin to hemoglobin (e.g., methemoglobin reductase, NADP diaphorase). Acquired methemoglobinemia is caused by toxins that oxidize heme iron, notably nitrate and nitrite-containing compounds, including drugs commonly used in cardiology and anesthesiology.
DIAGNOSIS AND MANAGEMENT OF PATIENTS WITH UNSTABLE HEMOGLOBINS, HIGH-AFFINITY HEMOGLOBINS, AND METHEMOGLOBINEMIA
Unstable hemoglobin variants should be suspected in patients with nonimmune hemolytic anemia, jaundice, splenomegaly, or premature biliary tract disease. Severe hemolysis usually presents during infancy as neonatal jaundice or anemia. Milder cases may present in adult life with anemia or only as unexplained reticulocytosis, hepatosplenomegaly, premature biliary tract disease, or leg ulcers. Because spontaneous mutation is common, family history of anemia may be absent. The peripheral blood smear often shows anisocytosis, abundant cells with punctate inclusions, and irregular shapes (i.e., poikilocytosis).
The two best tests for diagnosing unstable hemoglobins are the Heinz body preparation and the isopropanol or heat stability test. Many unstable Hb variants are electrophoretically silent. A normal electrophoresis does not rule out the diagnosis. Mass spectroscopy or direct gene analysis will provide a definitive diagnosis.
Severely affected patients may require transfusion support for the first 3 years of life, because splenectomy before age 3 is associated with a significantly higher immune deficit. Splenectomy is usually effective thereafter, but occasional patients may require lifelong transfusion support. After splenectomy, patients can develop cholelithiasis and leg ulcers, hypercoagulable states, and susceptibility to overwhelming sepsis. Splenectomy should thus be avoided or delayed unless it is the only alternative. Precipitation of unstable hemoglobins is aggravated by oxidative stress, e.g., infection and antimalarial drugs, which should be avoided where possible.
High-O2 affinity hemoglobin variants should be suspected in patients with erythrocytosis. The best test for confirmation is measurement of the P50. A high-O2 affinity hemoglobin causes a significant left shift (i.e., lower numeric value of the P50); confounding conditions, e.g., tobacco smoking or carbon monoxide exposure, can also lower the P50.
High-affinity hemoglobins are often asymptomatic; rubor or plethora may be telltale signs. When the hematocrit approaches 60%, symptoms of high blood viscosity and sluggish flow (headache, lethargy, dizziness, etc.) may be present. These persons may benefit from judicious phlebotomy. Erythrocytosis represents an appropriate attempt to compensate for the impaired oxygen delivery by the abnormal variant. Overzealous phlebotomy may stimulate increased erythropoiesis or aggravate symptoms by thwarting this compensatory mechanism. The guiding principle of phlebotomy should be to improve oxygen delivery by reducing blood viscosity and increasing blood flow rather than restoration of a normal hematocrit. Phlebotomy-induced modest iron deficiency may aid in control.
Low-affinity hemoglobins should be considered in patients with cyanosis or a low hematocrit with no other reason apparent after thorough evaluation. The P50 test confirms the diagnosis. Counseling and reassurance are the interventions of choice.
Methemoglobin should be suspected in patients with hypoxic symptoms who appear cyanotic but have a Pao2 sufficiently high that hemoglobin should be fully saturated with oxygen. A history of nitrite or other oxidant ingestions may not always be available; some exposures may be inapparent to the patient, and others may result from suicide attempts. The characteristic muddy appearance of freshly drawn blood can be a critical clue. The best diagnostic test is methemoglobin assay, which is usually available on an emergency basis.
Methemoglobinemia often causes symptoms of cerebral ischemia at levels >15%; levels >60% are usually lethal. Intravenous injection of 1 mg/kg of methylene blue is effective emergency therapy. Milder cases and follow-up of severe cases can be treated orally with methylene blue (60 mg three to four times each day) or ascorbic acid (300–600 mg/d).
THALASSEMIA SYNDROMES
The thalassemia syndromes are inherited disorders of α- or β-globin biosynthesis. The reduced supply of globin diminishes production of hemoglobin tetramers, causing hypochromia and microcytosis. Unbalanced accumulation of α and β subunits occurs because the synthesis of the unaffected globins proceeds at a normal rate. Unbalanced chain accumulation dominates the clinical phenotype. Clinical severity varies widely, depending on the degree to which the synthesis of the affected globin is impaired, altered synthesis of other globin chains, and coinheritance of other abnormal globin alleles.
CLINICAL MANIFESTATIONS OF β THALASSEMIA SYNDROMES
Mutations causing thalassemia can affect any step in the pathway of globin gene expression: transcription, processing of the mRNA precursor, translation, and posttranslational metabolism of the β-globin polypeptide chain. The most common forms arise from mutations that derange splicing of the mRNA precursor or prematurely terminate translation of the mRNA.
Hypochromia and microcytosis characterize all forms of β thalassemia because of the reduced amounts of hemoglobin tetramers (Fig. 127-5). In heterozygotes (β thalassemia trait), this is the only abnormality seen. Anemia is minimal. In more severe homozygous states, unbalanced α- and β-globin accumulation causes accumulation of highly insoluble unpaired α chains. They form toxic inclusion bodies that kill developing erythroblasts in the marrow. Few of the proerythroblasts beginning erythroid maturation survive. The surviving RBCs bear a burden of inclusion bodies that are detected in the spleen, shortening the RBC life span and producing severe hemolytic anemia. The resulting profound anemia stimulates erythropoietin release and compensatory erythroid hyperplasia, but the marrow response is sabotaged by the ineffective erythropoiesis. Anemia persists. Erythroid hyperplasia can become exuberant and produce masses of extramedullary erythropoietic tissue in the liver and spleen.
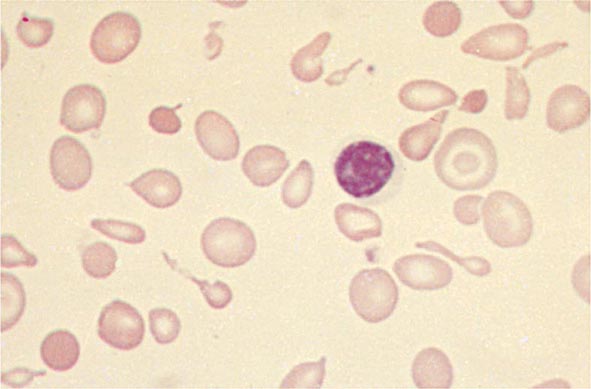
FIGURE 127-5 β Thalassemia intermedia. Microcytic and hypochromic red blood cells are seen that resemble the red blood cells of severe iron-deficiency anemia. Many elliptical and teardrop-shaped red blood cells are noted.
Massive bone marrow expansion deranges growth and development. Children develop characteristic “chipmunk” facies due to maxillary marrow hyperplasia and frontal bossing. Thinning and pathologic fracture of long bones and vertebrae may occur due to cortical invasion by erythroid elements and profound growth retardation. Hemolytic anemia causes hepatosplenomegaly, leg ulcers, gallstones, and high-output congestive heart failure. The conscription of caloric resources to support erythropoiesis leads to inanition, susceptibility to infection, endocrine dysfunction, and in the most severe cases, death during the first decade of life. Chronic transfusions with RBCs improve oxygen delivery, suppress the excessive ineffective erythropoiesis, and prolong life, but the inevitable side effects, notably iron overload, often prove fatal by age 30 years.
Severity is highly variable. Known modulating factors are those that ameliorate the burden of unpaired α-globin inclusions. Alleles associated with milder synthetic defects and coinheritance of α thalassemia trait reduce clinical severity by reducing accumulation of excess α globin. HbF persists to various degrees in β thalassemias. γ-Globin gene chains can substitute for β chains, generating more hemoglobin and reducing the burden of α-globin inclusions. The terms β thalassemia major and β thalassemia intermedia are used to reflect the clinical heterogeneity. Patients with β thalassemia major require intensive transfusion support to survive. Patients with β thalassemia intermedia have a somewhat milder phenotype and can survive without transfusion. The terms β thalassemia minor and β thalassemia trait describe asymptomatic heterozygotes for β thalassemia.
THALASSEMIA SYNDROMES
The four classic α thalassemias, most common in Asians, are α thalassemia-2 trait, in which one of the four α-globin loci is deleted; α thalassemia-1 trait, with two deleted loci; HbH disease, with three loci deleted; and hydrops fetalis with Hb Barts, with all four loci deleted (Table 127-4). Nondeletion forms of α thalassemia also exist.
THE α THALASSEMIAS |
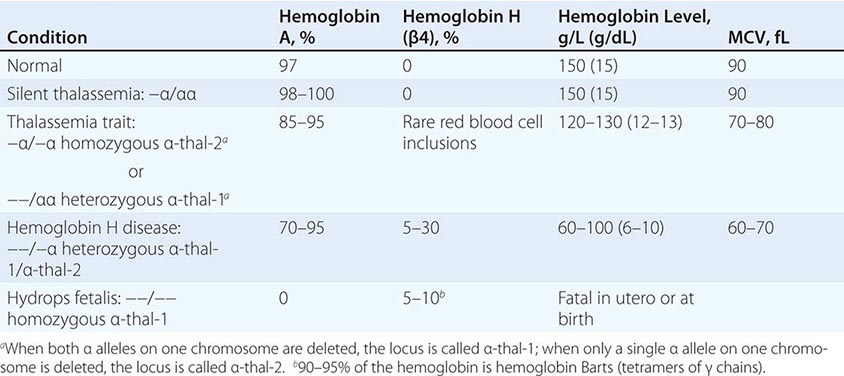
α Thalassemia-2 trait is an asymptomatic, silent carrier state. α Thalassemia-1 trait resembles β thalassemia minor. Offspring doubly heterozygous for α thalassemia-2 and α thalassemia-1 exhibit a more severe phenotype called HbH disease. Heterozygosity for a deletion that removes both genes from the same chromosome (cis deletion) is common in Asians and in those from the Mediterranean region, as is homozygosity for α thalassemia-2 (trans deletion). Both produce asymptomatic hypochromia and microcytosis.
In HbH disease, HbA production is only 25–30% normal. Fetuses accumulate some unpaired γ chains (Hb Barts; γ-chain tetramers). In adults, unpaired β chains accumulate and are soluble enough to form β4 tetramers called HbH. HbH forms few inclusions in erythroblasts and precipitates in circulating RBC. Patients with HbH disease have thalassemia intermedia characterized by moderately severe hemolytic anemia but milder ineffective erythropoiesis. Survival into midadult life without transfusions is common.
The homozygous state for the α thalassemia-1 cis deletion (hydrops fetalis) causes total absence of α-globin synthesis. No physiologically useful hemoglobin is produced beyond the embryonic stage. Excess γ globin forms tetramers called Hb Barts (γ4), which has a very high oxygen affinity. It delivers almost no O2 to fetal tissues, causing tissue asphyxia, edema (hydrops fetalis), congestive heart failure, and death in utero. α Thalassemia-2 trait is common (15–20%) among people of African descent. The cis α thalassemia-1 deletion is almost never seen, however. Thus, α thalassemia-2 and the trans form of α thalassemia-1 are very common, but HbH disease and hydrops fetalis are rare.
It has been known for some time that some patients with myelodysplasia or erythroleukemia produce RBC clones containing HbH. This phenomenon is due to mutations in the ATRX pathway that affect the LCR of the α-globin gene cluster.
DIAGNOSIS AND MANAGEMENT OF THALASSEMIAS
The diagnosis of β-thalassemia major is readily made during childhood on the basis of severe anemia accompanied by the characteristic signs of massive ineffective erythropoiesis: hepatosplenomegaly, profound microcytosis, a characteristic blood smear (Fig. 127-5), and elevated levels of HbF, HbA2, or both. Many patients require chronic hypertransfusion therapy designed to maintain a hematocrit of at least 27–30% so that erythropoiesis is suppressed. Splenectomy is required if the annual transfusion requirement (volume of RBCs per kilogram of body weight per year) increases by >50%. Folic acid supplements may be useful. Vaccination with Pneumovax in anticipation of eventual splenectomy is advised, as is close monitoring for infection, leg ulcers, and biliary tract disease. Many patients develop endocrine deficiencies as a result of iron overload. Early endocrine evaluation is required for glucose intolerance, thyroid dysfunction, and delayed onset of puberty or secondary sexual characteristics.
Patients with β thalassemia intermedia exhibit similar stigmata but can survive without chronic hypertransfusion. Management is particularly challenging because a number of factors can aggravate the anemia, including infection, onset of puberty, and development of splenomegaly and hypersplenism. Some patients may eventually benefit from splenectomy. The expanded erythron can cause absorption of excessive dietary iron and hemosiderosis, even without transfusion. Some patients eventually become transfusion dependent.
β Thalassemia minor (i.e., thalassemia trait) usually presents as profound microcytosis and hypochromia with target cells, but only minimal or mild anemia. The mean corpuscular volume is rarely >75 fL; the hematocrit is rarely <30–33%. Hemoglobin analysis classically reveals an elevated HbA2 (3.5–7.5%), but some forms are associated with normal HbA2 and/or elevated HbF. Genetic counseling and patient education are essential. Patients with β thalassemia trait should be warned that their blood picture resembles iron deficiency and can be misdiagnosed. They should eschew empirical use of iron, yet iron deficiency requiring replacement therapy can develop during pregnancy or from chronic bleeding.
Persons with α thalassemia trait may exhibit mild hypochromia and microcytosis usually without anemia. HbA2 and HbF levels are normal. Affected individuals usually require only genetic counseling. HbH disease resembles β thalassemia intermedia, with the added complication that the HbH molecule behaves like moderately unstable hemoglobin. Patients with HbH disease should undergo splenectomy if excessive anemia or a transfusion requirement develops. Oxidative drugs should be avoided. Iron overload leading to death can occur in more severely affected patients.
PREVENTION
Antenatal diagnosis of thalassemia syndromes is now widely available. DNA diagnosis is based on polymerase chain reaction (PCR) amplification of fetal DNA, obtained by amniocentesis or chorionic villus biopsy followed by hybridization to allele-specific oligonucleotide probes or direct DNA sequencing.
THALASSEMIC STRUCTURAL VARIANTS
Thalassemic structural variants are characterized by both defective synthesis and abnormal structure.
HEMOGLOBIN LEPORE
Hb Lepore [α2(δβ)2] arises by an unequal crossover and recombination event that fuses the proximal end of the δ-gene with the distal end of the closely linked β-gene. It is common in the Mediterranean basin. The resulting chromosome contains only the fused δβ gene. The Lepore (δβ) globin is synthesized poorly because the fused gene is under the control of the weak δ-globin promoter. Hb Lepore alleles have a phenotype like β thalassemia, except for the added presence of 2–20% Hb Lepore. Compound heterozygotes for Hb Lepore and a classic β thalassemia allele may also have severe thalassemia.
HEMOGLOBIN E
![]() HbE (i.e., α2β226Glu→Lys) is extremely common in Cambodia, Thailand, and Vietnam. The gene has become far more prevalent in the United States as a result of immigration of Asian persons, especially in California, where HbE is the most common variant detected. HbE is mildly unstable but not enough to affect RBC life span significantly. Heterozygotes resemble individuals with a mild β-thalassemia trait. Homozygotes have somewhat more marked abnormalities but are asymptomatic. Compound heterozygotes for HbE and a β thalassemia gene can have β thalassemia intermedia or β thalassemia major, depending on the severity of the coinherited thalassemic gene.
HbE (i.e., α2β226Glu→Lys) is extremely common in Cambodia, Thailand, and Vietnam. The gene has become far more prevalent in the United States as a result of immigration of Asian persons, especially in California, where HbE is the most common variant detected. HbE is mildly unstable but not enough to affect RBC life span significantly. Heterozygotes resemble individuals with a mild β-thalassemia trait. Homozygotes have somewhat more marked abnormalities but are asymptomatic. Compound heterozygotes for HbE and a β thalassemia gene can have β thalassemia intermedia or β thalassemia major, depending on the severity of the coinherited thalassemic gene.
The βE allele contains a single base change in codon 26 that causes the amino acid substitution. This mutation also activates a cryptic RNA splice site, generating a structurally abnormal globin mRNA that cannot be translated, from about 50% of the initial pre-mRNA molecules. The remaining 40–50% are normally spliced and generate functional mRNA that is translated into βE-globin because the mature mRNA carries the base change that alters codon 26.
Genetic counseling of the persons at risk for HbE should focus especially on the interaction of HbE with β thalassemia, because HbE homozygosity is a condition associated with mildly asymptomatic microcytosis, hypochromia, and hemoglobin levels rarely <100 g/L (<10 g/dL).
HEREDITARY PERSISTENCE OF FETAL HEMOGLOBIN
HPFH is characterized by continued synthesis of high levels of HbF in adult life. No deleterious effects are apparent, even when all of the hemoglobin produced is HbF. These rare patients demonstrate convincingly that prevention or reversal of the fetal to adult hemoglobin switch would provide effective therapy for sickle cell anemia and β thalassemia.
ACQUIRED HEMOGLOBINOPATHIES
The two most important acquired hemoglobinopathies are carbon monoxide poisoning and methemoglobinemia (see above). Carbon monoxide has a higher affinity for hemoglobin than does oxygen; it can replace oxygen and diminish O2 delivery. Chronic elevation of carboxyhemoglobin levels to 10 or 15%, as occurs in smokers, can lead to secondary polycythemia. Carboxyhemoglobin is cherry red in color and masks the development of cyanosis usually associated with poor O2 delivery to tissues.
Abnormalities of hemoglobin biosynthesis have also been described in blood dyscrasias. In some patients with myelodysplasia, erythroleukemia, or myeloproliferative disorders, elevated HbF or a mild form of HbH disease may also be seen. The abnormalities are not severe enough to alter the course of the underlying disease.
EXPERIMENTAL THERAPIES
BONE MARROW TRANSPLANTATION, GENE THERAPY, AND MANIPULATION OF HbF
Bone marrow transplantation provides stem cells able to express normal hemoglobin; it has been used in a large number of patients with β thalassemia and a smaller number of patients with sickle cell anemia. Early in the course of disease, before end-organ damage occurs, transplantation is curative in 80–90% of patients. In highly experienced centers, the treatment-related mortality is <10%. Because survival into adult life is possible with conventional therapy, the decision to transplant is best made in consultation with specialized centers.
Gene therapy of thalassemia and sickle cell disease has proved to be an elusive goal, but experimental advances are raising expectations.
Reestablishing high levels of fetal hemoglobin synthesis should ameliorate the symptoms of β-chain hemoglobinopathies. Cytotoxic agents such as hydroxyurea and cytarabine promote high levels of HbF synthesis, probably by stimulating proliferation of the primitive HbF-producing progenitor cell population (i.e., F cell progenitors). Unfortunately, this regimen has not yet been effective in β thalassemia. Butyrates stimulate HbF production, but only transiently. Pulsed or intermittent administration has been found to sustain HbF induction in the majority of patients with sickle cell disease. It is unclear whether butyrates will have similar activity in patients with β thalassemia.
APLASTIC AND HYPOPLASTIC CRISIS IN PATIENTS WITH HEMOGLOBINOPATHIES
Patients with hemolytic anemias sometimes exhibit an alarming decline in hematocrit during and immediately after acute illnesses. Bone marrow suppression occurs in almost everyone during acute and chronic inflammatory illnesses. In patients with short RBC life spans, suppression can affect RBC counts more dramatically. These hypoplastic crises are usually transient and self-correcting before intervention is required.
Aplastic crisis refers to a profound cessation of erythroid activity in patients with chronic hemolytic anemias. It is associated with a rapidly falling hematocrit. Episodes are usually self-limited. Aplastic crises are caused by infection with a particular strain of parvovirus, B19A. Children infected with this virus usually develop permanent immunity. Aplastic crises do not often recur and are rarely seen in adults. Management requires close monitoring of the hematocrit and reticulocyte count. If anemia becomes symptomatic, transfusion support is indicated. Most crises resolve spontaneously within 1–2 weeks.
128 | Megaloblastic Anemias |
The megaloblastic anemias are a group of disorders characterized by the presence of distinctive morphologic appearances of the developing red cells in the bone marrow. The marrow is usually hypercellular and the anemia is based on ineffective erythropoiesis. The cause is usually a deficiency of either cobalamin (vitamin B12) or folate, but megaloblastic anemia may occur because of genetic or acquired abnormalities that affect the metabolism of these vitamins or because of defects in DNA synthesis not related to cobalamin or folate (Table 128-1). Cobalamin and folate absorption and metabolism are described next, followed by the biochemical basis, clinical and laboratory features, causes, and treatment of megaloblastic anemia.
CAUSES OF MEGALOBLASTIC ANEMIA |
COBALAMIN
Cobalamin (vitamin B12) exists in a number of different chemical forms. All have a cobalt atom at the center of a corrin ring. In nature, the vitamin is mainly in the 2-deoxyadenosyl (ado) form, which is located in mitochondria. It is the cofactor for the enzyme methylmalonyl coenzyme A (CoA) mutase. The other major natural cobalamin is methylcobalamin, the form in human plasma and in cell cytoplasm. It is the cofactor for methionine synthase. There are also minor amounts of hydroxocobalamin to which methyl- and adocobalamin are converted rapidly by exposure to light.
DIETARY SOURCES AND REQUIREMENTS
Cobalamin is synthesized solely by microorganisms. Ruminants obtain cobalamin from the foregut, but the only source for humans is food of animal origin, e.g., meat, fish, and dairy products. Vegetables, fruits, and other foods of nonanimal origin are free from cobalamin unless they are contaminated by bacteria. A normal Western diet contains 5–30 μg of cobalamin daily. Adult daily losses (mainly in the urine and feces) are 1–3 μg (~0.1% of body stores), and because the body does not have the ability to degrade cobalamin, daily requirements are also about 1–3 μg. Body stores are of the order of 2–3 mg, sufficient for 3–4 years if supplies are completely cut off.
ABSORPTION
Two mechanisms exist for cobalamin absorption. One is passive, occurring equally through buccal, duodenal, and ileal mucosa; it is rapid but extremely inefficient, with <1% of an oral dose being absorbed by this process. The normal physiologic mechanism is active; it occurs through the ileum and is efficient for small (a few micrograms) oral doses of cobalamin, and it is mediated by gastric intrinsic factor (IF). Dietary cobalamin is released from protein complexes by enzymes in the stomach, duodenum, and jejunum; it combines rapidly with a salivary glycoprotein that belongs to the family of cobalamin-binding proteins known as haptocorrins (HCs). In the intestine, the haptocorrin is digested by pancreatic trypsin and the cobalamin is transferred to IF.
IF (gene at chromosome 11q13) is produced in the gastric parietal cells of the fundus and body of the stomach, and its secretion parallels that of hydrochloric acid. Normally, there is a vast excess of IF. The IF-cobalamin complex passes to the ileum, where IF attaches to a specific receptor (cubilin) on the microvillus membrane of the enterocytes. Cubilin also is present in yolk sac and renal proximal tubular epithelium. Cubilin appears to traffic by means of amnionless (AMN), an endocytic receptor protein that directs sublocalization and endocytosis of cubilin with its ligand IF-cobalamin complex. The cobalamin-IF complex enters the ileal cell, where IF is destroyed. After a delay of about 6 h, the cobalamin appears in portal blood attached to transcobalamin (TC) II.
Between 0.5 and 5 μg of cobalamin enter the bile each day. This binds to IF, and a major portion of biliary cobalamin normally is reabsorbed together with cobalamin derived from sloughed intestinal cells. Because of the appreciable amount of cobalamin undergoing enterohepatic circulation, cobalamin deficiency develops more rapidly in individuals who malabsorb cobalamin than it does in vegans, in whom reabsorption of biliary cobalamin is intact.
TRANSPORT
Two main cobalamin transport proteins exist in human plasma; they both bind cobalamin—one molecule for one molecule. One HC, also known as TC I, is closely related to other cobalamin-binding HCs in milk, gastric juice, bile, saliva, and other fluids. The gene TCNL is at chromosome 11q11-q12.3. These HCs differ from each other only in the carbohydrate moiety of the molecule. TC I is derived primarily from the specific granules in neutrophils. Normally, it is about two-thirds saturated with cobalamin, which it binds tightly. TC I does not enhance cobalamin entry into tissues. Glycoprotein receptors on liver cells are involved in the removal of TC I from plasma, and TC I may play a role in the transport of cobalamin analogues (which it binds more effectively than IF) to the liver for excretion in bile.
The other major cobalamin transport protein in plasma is transcobalamin, also known as TC II. The gene is on chromosome 22q11-q13.1. As for IF and HC, there are nine exons. The three proteins are likely to have a common ancestral origin. TC II is synthesized by liver and by other tissues, including macrophages, ileum, and vascular endothelium. It normally carries only 20–60 ng of cobalamin per liter of plasma and readily gives up cobalamin to marrow, placenta, and other tissues, which it enters by receptor-mediated endocytosis involving the TC II receptor and megalin (encoded by the LRP-2 gene). The TC II cobalamin is internalized by endocytosis via clathrin-coated pits; the complex is degraded, but the receptor probably is recycled to the cell membrane as is the case for transferrin. Export of “free” cobalamin is via the ATP-binding cassette drug transporter alias multidrug resistance protein 1.
FOLATE
DIETARY FOLATE
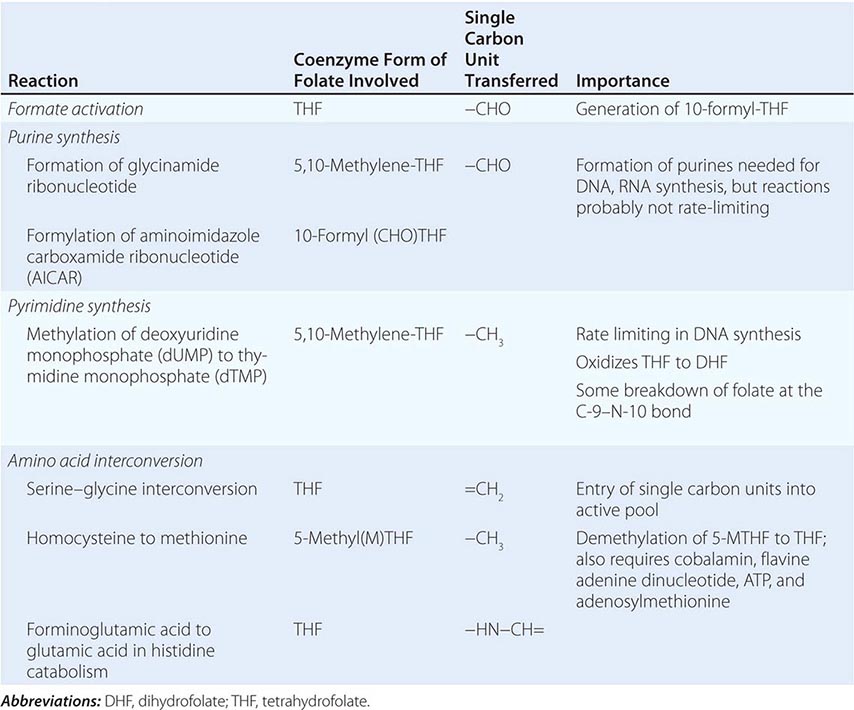
Folic (pteroylglutamic) acid is a yellow, crystalline, water-soluble substance. It is the parent compound of a large family of natural folate compounds, which differ from it in three respects: (1) they are partly or completely reduced to di- or tetrahydrofolate (THF) derivatives, (2) they usually contain a single carbon unit (Table 128-2), and (3) 70–90% of natural folates are folate-polyglutamates.
BIOCHEMICAL REACTIONS OF FOLATE COENZYMES |
Most foods contain some folate. The highest concentrations are found in liver, yeast, spinach, other greens, and nuts (>100 μg/100 g). The total folate content of an average Western diet is ~250 μg daily, but the amount varies widely according to the type of food eaten and the method of cooking. Folate is easily destroyed by heating, particularly in large volumes of water. Total body folate in the adult is ~10 mg, with the liver containing the largest store. Daily adult requirements are ~100 μg, and so stores are sufficient for only 3–4 months in normal adults and severe folate deficiency may develop rapidly.
ABSORPTION
Folates are absorbed rapidly from the upper small intestine. The absorption of folate polyglutamates is less efficient than that of monoglutamates; on average, ~50% of food folate is absorbed. Polyglutamate forms are hydrolyzed to the monoglutamate derivatives either in the lumen of the intestine or within the mucosa. All dietary folates are converted to 5-methylTHF (5-MTHF) within the small intestinal mucosa before entering portal plasma. The monoglutamates are actively transported across the enterocyte by a proton-coupled folate transporter (PCFT, SCL46A1). This is situated at the apical brush border and is most active at pH 5.5, which is about the pH of the duodenal and jejunal surface. Genetic mutations of this protein underlie hereditary malabsorption of folate (see below). Pteroylglutamic acid at doses >400 μg is absorbed largely unchanged and converted to natural folates in the liver. Lower doses are converted to 5-MTHF during absorption through the intestine.
About 60–90 μg of folate enters the bile each day and is excreted into the small intestine. Loss of this folate, together with the folate of sloughed intestinal cells, accelerates the speed with which folate deficiency develops in malabsorption conditions.
TRANSPORT
Folate is transported in plasma; about one-third is loosely bound to albumin, and two-thirds is unbound. In all body fluids (plasma, cerebrospinal fluid, milk, bile), folate is largely, if not entirely, 5-MTHF in the monoglutamate form. Three types of folate-binding protein are involved. A reduced folate transporter (RFC, SLC19A1) is the major route of delivery of plasma folate (5-MTHF) to cells. Two folate receptors, FR2 and FR3 embedded in the cell membrane by a glycosyl phosphatidylinositol anchor, transport folate into the cell via receptor-mediated endocytosis. The third protein, PCFT, transports folate at low pH from the vesicle to the cell cytoplasm. The reduced folate transporter also mediates uptake of methotrexate by cells.
BIOCHEMICAL FUNCTIONS
Folates (as the intracellular polyglutamate derivatives) act as coenzymes in the transfer of single-carbon units (Fig. 128-1 and Table 128-2). Two of these reactions are involved in purine synthesis and one in pyrimidine synthesis necessary for DNA and RNA replication. Folate is also a coenzyme for methionine synthesis, in which methylcobalamin is also involved and in which THF is regenerated. THF is the acceptor of single carbon units newly entering the active pool via conversion of serine to glycine. Methionine, the other product of the methionine synthase reaction, is the precursor for S-adenosylmethionine (SAM), the universal methyl donor involved in >100 methyltransferase reactions (Fig. 128-1).
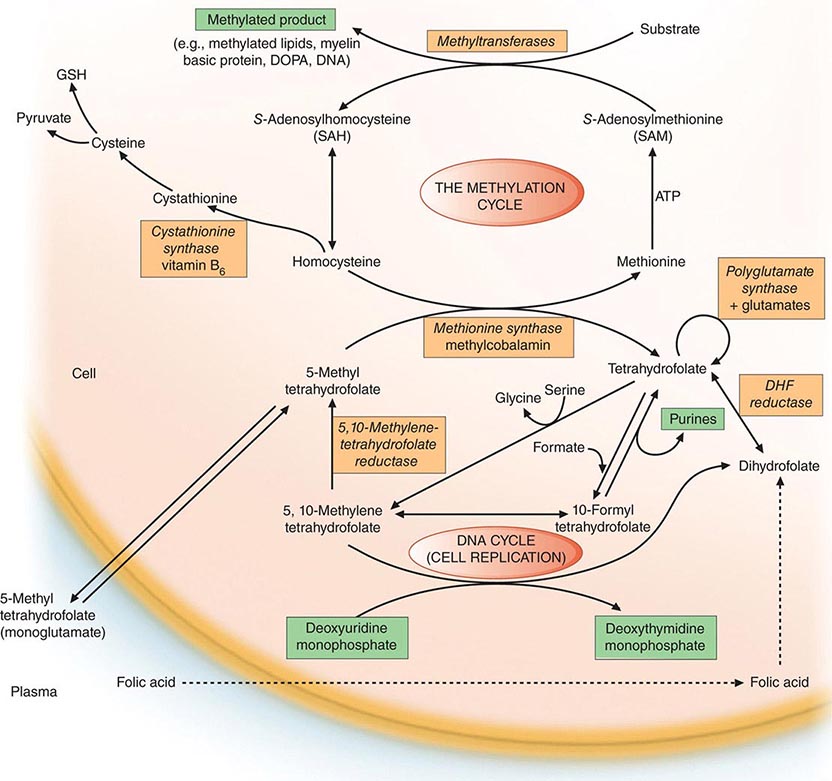
FIGURE 128-1 The role of folates in DNA synthesis and in formation of S-adenosylmethionine (SAM), which is involved in numerous methylation reactions. DHF, dihydrofolate; GSH, glutathione. (Reprinted from AV Hoffbrand et al [eds]: Postgraduate Haematology, 5th ed. Oxford, UK, Blackwell Publishing, 2005; with permission.)
During thymidylate synthesis, 5,10-methylene-THF is oxidized to DHF (dihydrofolate). The enzyme DHF reductase converts this to THF. The drugs methotrexate, pyrimethamine, and (mainly in bacteria) trimethoprim inhibit DHF reductase and so prevent formation of active THF coenzymes from DHF. A small fraction of the folate coenzyme is not recycled during thymidylate synthesis but is degraded at the C9-N10 bond.
BIOCHEMICAL BASIS OF MEGALOBLASTIC ANEMIA
The common feature of all megaloblastic anemias is a defect in DNA synthesis that affects rapidly dividing cells in the bone marrow. All conditions that give rise to megaloblastic changes have in common a disparity in the rate of synthesis or availability of the four immediate precursors of DNA: the deoxyribonucleoside triphosphates (dNTPs)—dA(adenine)TP and dG(guanine)TP (purines), dT(thymine)TP and dC(cytosine)TP (pyrimidines). In deficiencies of either folate or cobalamin, there is failure to convert deoxyuridine monophosphate (dUMP) to deoxythymidine monophosphate (dTMP), the precursor of dTTP (Fig 128-1). This is the case because folate is needed as the coenzyme 5,10-methylene-THF polyglutamate for conversion of dUMP to dTMP; the availability of 5,10-methylene-THF is reduced in either cobalamin or folate deficiency. An alternative theory for megaloblastic anemia in cobalamin or folate deficiency is misincorporation of uracil into DNA because of a buildup of deoxyuridine triphosphate (dUTP) at the DNA replication fork as a consequence of the block in conversion of dUMP to dTMP.
COBALAMIN-FOLATE RELATIONS
Folate is required for many reactions in mammalian tissues. Only two reactions in the body are known to require cobalamin. Methylmalonyl CoA isomerization requires adocobalamin, and the methylation of homocysteine to methionine requires both methylcobalamin and 5-MTHF (Fig. 128-1). This reaction is the first step in the pathway by which 5-MTHF, which enters bone marrow and other cells from plasma, is converted into all the intracellular folate coenzymes. The coenzymes are all polyglutamated (the larger size aiding retention in the cell), but the enzyme folate polyglutamate synthase can use only THF, not MTHF, as substrate. In cobalamin deficiency, MTHF accumulates in plasma, and intracellular folate concentrations fall due to failure of formation of THF, the substrate on which folate polyglutamates are built. This has been termed THF starvation, or the methylfolate trap.
This theory explains the abnormalities of folate metabolism that occur in cobalamin deficiency (high serum folate, low cell folate, positive purine precursor aminoimidazole carboxamide ribonucleotide [AICAR] excretion; Table 128-2) and also why the anemia of cobalamin deficiency responds to folic acid in large doses.
CLINICAL FEATURES
Many symptomless patients are detected through the finding of a raised mean corpuscular volume (MCV) on a routine blood count. The main clinical features in more severe cases are those of anemia. Anorexia is usually marked, and there may be weight loss, diarrhea, or constipation. Glossitis, angular cheilosis, a mild fever in more severely anemic patients, jaundice (unconjugated), and reversible melanin skin hyperpigmentation also may occur with a deficiency of either folate or cobalamin. Thrombocytopenia sometimes leads to bruising, and this may be aggravated by vitamin C deficiency or alcohol in malnourished patients. The anemia and low leukocyte count may predispose to infections, particularly of the respiratory and urinary tracts. Cobalamin deficiency has also been associated with impaired bactericidal function of phagocytes.
GENERAL TISSUE EFFECTS OF COBALAMIN AND FOLATE DEFICIENCIES
Epithelial Surfaces After the marrow, the next most frequently affected tissues are the epithelial cell surfaces of the mouth, stomach, and small intestine and the respiratory, urinary, and female genital tracts. The cells show macrocytosis, with increased numbers of multinucleate and dying cells. The deficiencies may cause cervical smear abnormalities.
Complications of Pregnancy The gonads are also affected, and infertility is common in both men and women with either deficiency. Maternal folate deficiency has been implicated as a cause of prematurity, and both folate deficiency and cobalamin deficiency have been implicated in recurrent fetal loss and neural tube defects, as discussed below.
Neural Tube Defects Folic acid supplements at the time of conception and in the first 12 weeks of pregnancy reduce by ~70% the incidence of neural tube defects (NTDs) (anencephaly, meningomyelocele, encephalocele, and spina bifida) in the fetus. Most of this protective effect can be achieved by taking folic acid, 0.4 mg daily, at the time of conception.
The incidence of cleft palate and harelip also can be reduced by prophylactic folic acid. There is no clear simple relationship between maternal folate status and these fetal abnormalities, although overall the lower the maternal folate, the greater the risk to the fetus. NTDs also can be caused by antifolate and antiepileptic drugs.
An underlying maternal folate metabolic abnormality has also been postulated. One abnormality has been identified: reduced activity of the enzyme 5,10-methylene-THF reductase (MTHFR) (Fig. 128-1) caused by a common C677T polymorphism in the MTHFR gene. In one study, the prevalence of this polymorphism was found to be higher than in controls in the parents of NTD fetuses and in the fetuses themselves: homozygosity for the TT mutation was found in 13% of cases compared with 5% of control subjects. The polymorphism codes for a thermolabile form of MTHFR. The homozygous state results in a lower mean serum and red cell folate level compared with control subjects, as well as significantly higher serum homocysteine levels. Tests for mutations in other enzymes possibly associated with NTDs, e.g., methionine synthase and serine–glycine hydroxymethylase, have been negative. Serum vitamin B12 levels are also lower in the sera of mothers of NTD infants than in controls. In addition, maternal TC II receptor polymorphisms are associated with increased risk of NTD births. There are, however, no studies showing dietary fortification with vitamin B12 reduces the incidence of NTDs.
Cardiovascular Disease Children with severe homocystinuria (blood levels ≥100 μmol/L) due to deficiency of one of three enzymes, methionine synthase, MTHFR, or cystathionine synthase (Fig. 128-1), have vascular disease, e.g., ischemic heart disease, cerebrovascular disease, or pulmonary embolus, as teenagers or in young adulthood. Lesser degrees of raised serum homocysteine and low levels of serum folate and homozygous inherited mutations of MTHFR have been found to be associated with cerebrovascular, peripheral vascular, and coronary heart disease and with deep vein thrombosis. Prospective randomized trials of lowering homocysteine levels with supplements of folic acid, vitamin B12, and vitamin B6 against placebo over a 5-year period in patients with vascular disease or diabetes have not, however, shown a reduction of first event fatal or nonfatal myocardial infarction, nor have these supplements reduced the risk of recurrent cardiovascular disease after an acute myocardial infarct. Meta-analysis showed an 18% reduction in strokes but no significant prevention of death from any cause. Venous thrombosis has been reported to be more frequent in vitamin B12–deficient subjects than in controls. This was ascribed to raised plasma homocysteine levels in vitamin B12 deficiency.
Malignancy Prophylactic folic acid in pregnancy has been found in some but not all studies to reduce the subsequent incidence of acute lymphoblastic leukemia (ALL) in childhood. A significant negative association has also been found with the MTHFR C677T polymorphism and leukemias with mixed lineage leukemia (MLL) translocations, but a positive association with hyperdiploidy in infants with ALL or acute myeloid leukemia or with childhood ALL. A second polymorphism in the MTHFR gene, A1298C, is also strongly associated with hyperdiploid leukemia. There are various positive and negative associations between polymorphisms in folate-dependent enzymes and the incidence of adult ALL. The C677T polymorphism is thought to lead to increased thymidine pools and “better quality” of DNA synthesis by shunting one-carbon groups toward thymidine and purine synthesis. This may explain its reported association with a lower risk for colorectal cancer. Most but not all studies suggest that prophylactic folic acid also protects against colon adenomas. Other tumors that have been associated with folate polymorphisms or status include follicular lymphoma, breast cancer, and gastric cancer. A meta-analysis of 50,000 individuals given folic acid or placebo in cardiovascular or colon adenoma prevention trials found that folic acid supplementation did not substantially increase or decrease the incidence of site-specific cancer during the first 5 years of treatment. Because folic acid may “feed” tumors, it probably should be avoided in those with established tumors unless there is severe megaloblastic anemia due to folate deficiency.
Neurologic Manifestations Cobalamin deficiency may cause a bilateral peripheral neuropathy or degeneration (demyelination) of the posterior and pyramidal tracts of the spinal cord and, less frequently, optic atrophy or cerebral symptoms.
The patient, more frequently male, presents with paresthesias, muscle weakness, or difficulty in walking and sometimes dementia, psychotic disturbances, or visual impairment. Long-term nutritional cobalamin deficiency in infancy leads to poor brain development and impaired intellectual development. Folate deficiency has been suggested to cause organic nervous disease, but this is uncertain, although methotrexate injected into the cerebrospinal fluid may cause brain or spinal cord damage.
An important clinical problem is the nonanemic patient with neurologic or psychiatric abnormalities and a low or borderline serum cobalamin level. In such patients, it is necessary to try to establish whether there is significant cobalamin deficiency, e.g., by careful examination of the blood film, tests for serum gastrin level and for antibodies to IF or parietal cells, along with serum methylmalonic acid (MMA) measurement if available. A trial of cobalamin therapy for at least 3 months will usually also be needed to determine whether the symptoms improve.
The biochemical basis for cobalamin neuropathy remains obscure. Its occurrence in the absence of methylmalonic aciduria in TC II deficiency suggests that the neuropathy is related to the defect in homocysteine-methionine conversion. Accumulation of S-adenosylhomocysteine in the brain, resulting in inhibition of transmethylation reactions, has been suggested.
Psychiatric disturbance is common in both folate and cobalamin deficiencies. This, like the neuropathy, has been attributed to a failure of the synthesis of SAM, which is needed in methylation of biogenic amines (e.g., dopamine) as well as that of proteins, phospholipids, and neurotransmitters in the brain (Fig. 128-1). Associations between lower serum folate or cobalamin levels and higher homocysteine levels and the development of decreased cognitive function and dementia in Alzheimer’s disease have been reported. A meta-analysis of randomized, placebo-controlled trials of homocysteine-lowering B-vitamin supplementation of individuals with and without cognitive impairment, however, showed that supplementation with vitamin B12, vitamin B6, and folic acid alone or in combination did not improve cognitive function. It is unknown whether prolonged treatment with these B vitamins can reduce the risk of dementia in later life.
HEMATOLOGIC FINDINGS
PERIPHERAL BLOOD
Oval macrocytes, usually with considerable anisocytosis and poikilocytosis, are the main feature (Fig. 128-2A). The MCV is usually >100 fL unless a cause of microcytosis (e.g., iron deficiency or thalassemia trait) is present. Some of the neutrophils are hypersegmented (more than five nuclear lobes). There may be leukopenia due to a reduction in granulocytes and lymphocytes, but this is usually >1.5 × 109/L; the platelet count may be moderately reduced, rarely to <40 × 109/L. The severity of all these changes parallels the degree of anemia. In a nonanemic patient, the presence of a few macrocytes and hypersegmented neutrophils in the peripheral blood may be the only indication of the underlying disorder.
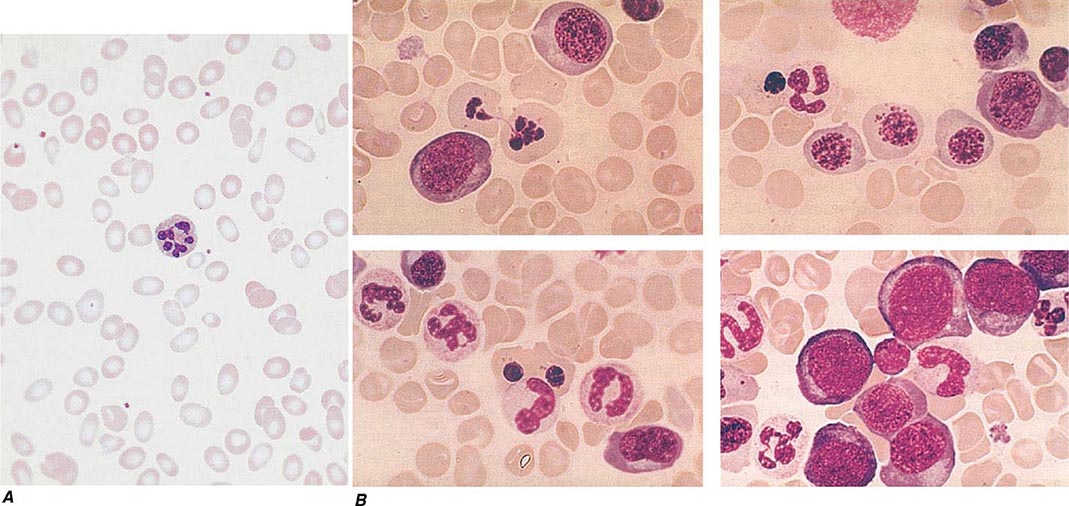
FIGURE 128-2 A. The peripheral blood in severe megaloblastic anemia. B. The bone marrow in severe megaloblastic anemia. (Reprinted from AV Hoffbrand et al [eds]: Postgraduate Haematology, 5th ed. Oxford, UK, Blackwell Publishing, 2005; with permission.)
BONE MARROW
In a severely anemic patient, the marrow is hypercellular with an accumulation of primitive cells due to selective death by apoptosis of more mature forms. The erythroblast nucleus maintains a primitive appearance despite maturation and hemoglobinization of the cytoplasm. The cells are larger than normoblasts, and an increased number of cells with eccentric lobulated nuclei or nuclear fragments may be present (Fig. 128-2B). Giant and abnormally shaped metamyelocytes and enlarged hyperpolyploid megakaryocytes are characteristic. In severe cases, the accumulation of primitive cells may mimic acute myeloid leukemia, whereas in less anemic patients, the changes in the marrow may be difficult to recognize. The terms intermediate, mild, and early have been used. The term megaloblastoid does not mean mildly megaloblastic. It is used to describe cells with both immature-appearing nuclei and defective hemoglobinization and is usually seen in myelodysplasia.
CHROMOSOMES
Bone marrow cells, transformed lymphocytes, and other proliferating cells in the body show a variety of changes, including random breaks, reduced contraction, spreading of the centromere, and exaggeration of secondary chromosomal constrictions and overprominent satellites. Similar abnormalities may be produced by antimetabolite drugs (e.g., cytosine arabinoside, hydroxyurea, and methotrexate) that interfere with either DNA replication or folate metabolism and that also cause megaloblastic appearances.
INEFFECTIVE HEMATOPOIESIS
There is an accumulation of unconjugated bilirubin in plasma due to the death of nucleated red cells in the marrow (ineffective erythropoiesis). Other evidence for this includes raised urine urobilinogen, reduced haptoglobins and positive urine hemosiderin, and a raised serum lactate dehydrogenase. A weakly positive direct antiglobulin test due to complement can lead to a false diagnosis of autoimmune hemolytic anemia.
CAUSES OF COBALAMIN DEFICIENCY
Cobalamin deficiency is usually due to malabsorption. The only other cause is inadequate dietary intake.
INADEQUATE DIETARY INTAKE
Adults Dietary cobalamin deficiency arises in vegans who omit meat, fish, eggs, cheese, and other animal products from their diet. The largest group in the world consists of Hindus, and it is likely that many millions of Indians are at risk of deficiency of cobalamin on a nutritional basis. Subnormal serum cobalamin levels are found in up to 50% of randomly selected, young, adult Indian vegans, but the deficiency usually does not progress to megaloblastic anemia since the diet of most vegans is not totally lacking in cobalamin and the enterohepatic circulation of cobalamin is intact. Dietary cobalamin deficiency may also arise rarely in nonvegetarian individuals who exist on grossly inadequate diets because of poverty or psychiatric disturbance.
Infants Cobalamin deficiency has been described in infants born to severely cobalamin-deficient mothers. These infants develop megaloblastic anemia at about 3–6 months of age, presumably because they are born with low stores of cobalamin and because they are fed breast milk with low cobalamin content. The babies have also shown growth retardation, impaired psychomotor development, and other neurologic sequelae.
GASTRIC CAUSES OF COBALAMIN MALABSORPTION
See Tables 128-3 and 128-4.
CAUSES OF COBALAMIN DEFICIENCY SUFFICIENTLY SEVERE TO CAUSE MEGALOBLASTIC ANEMIA |
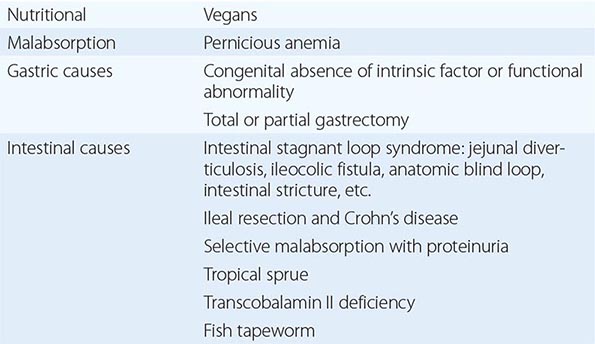
MALABSORPTION OF COBALAMIN MAY OCCUR IN THE FOLLOWING CONDITIONS BUT IS NOT USUALLY SUFFICIENTLY SEVERE AND PROLONGED TO CAUSE MEGALOBLASTIC ANEMIA |
Pernicious Anemia Pernicious anemia (PA) may be defined as a severe lack of IF due to gastric atrophy. It is a common disease in north Europeans but occurs in all countries and ethnic groups. The overall incidence is about 120 per 100,000 population in the United Kingdom (UK). The ratio of incidence in men and women among whites is ~1:1.6, and the peak age of onset is 60 years, with only 10% of patients being <40 years of age. However, in some ethnic groups, notably black individuals and Latin Americans, the age at onset of PA is generally lower. The disease occurs more commonly than by chance in close relatives and in persons with other organ-specific autoimmune diseases, e.g., thyroid diseases, vitiligo, hypoparathyroidism, and Addison’s disease. It is also associated with hypogammaglobulinemia, with premature graying or blue eyes, and persons of blood group A. An association with human leukocyte antigen (HLA) 3 has been reported in some but not all series and, in those with endocrine disease, with HLA-B8, -B12, and -BW15. Life expectancy is normal in women once regular treatment has begun. Men have a slightly subnormal life expectancy as a result of a higher incidence of carcinoma of the stomach than in control subjects. Gastric output of hydrochloric acid, pepsin, and IF is severely reduced. The serum gastrin level is raised, and serum pepsinogen I levels are low.
Gastric Biopsy A single endoscopic examination is recommended if PA is diagnosed. Gastric biopsy usually shows atrophy of all layers of the body and fundus, with loss of glandular elements, an absence of parietal and chief cells and replacement by mucous cells, a mixed inflammatory cell infiltrate, and perhaps intestinal metaplasia. The infiltrate of plasma cells and lymphocytes contains an excess of CD4 cells. These are directed against gastric H/K-ATPase. The antral mucosa is usually well preserved. Helicobacter pylori infection occurs infrequently in PA, but it has been suggested that H. pylori gastritis occurs at an early phase of atrophic gastritis and presents in younger patients as iron-deficiency anemia but in older patients as PA. H. pylori is suggested to stimulate an autoimmune process directed against parietal cells, with the H. pylori infection then being gradually replaced, in some individuals, by an autoimmune process.
Serum Antibodies Two types of IF immunoglobulin G antibody may be found in the sera of patients with PA. One, the “blocking,” or type I, antibody, prevents the combination of IF and cobalamin, whereas the “binding,” or type II, antibody prevents attachment of IF to ileal mucosa. Type I occurs in the sera of ~55% of patients, and type II in 35%. IF antibodies cross the placenta and may cause temporary IF deficiency in a newborn infant. Patients with PA also show cell-mediated immunity to IF. Type I antibody has been detected rarely in the sera of patients without PA but with thyrotoxicosis, myxedema, Hashimoto’s disease, or diabetes mellitus and in relatives of PA patients. IF antibodies also have been detected in gastric juice in ~80% of PA patients. These gastric antibodies may reduce absorption of dietary cobalamin by combining with small amounts of remaining IF.
Parietal cell antibody is present in the sera of almost 90% of adult patients with PA but is frequently present in other subjects. Thus, it occurs in as many as 16% of randomly selected female subjects age >60 years. The parietal cell antibody is directed against the α and β subunits of the gastric proton pump (H+,K+-ATPase).
JUVENILE PERNICIOUS ANEMIA
This usually occurs in older children and resembles PA of adults. Gastric atrophy, achlorhydria, and serum IF antibodies are all present, although parietal cell antibodies are usually absent. About one-half of these patients show an associated endocrinopathy such as autoimmune thyroiditis, Addison’s disease, or hypoparathyroidism; in some, mucocutaneous candidiasis occurs.
CONGENITAL INTRINSIC FACTOR DEFICIENCY OR FUNCTIONAL ABNORMALITY
An affected child usually presents with megaloblastic anemia in the first to third year of life; a few have presented as late as the second decade. The child usually has no demonstrable IF but has a normal gastric mucosa and normal secretion of acid. The inheritance is autosomal recessive. Parietal cell and IF antibodies are absent. Variants have been described in which the child is born with IF that can be detected immunologically but is unstable or functionally inactive, unable to bind cobalamin or to facilitate its uptake by ileal receptors.
GASTRECTOMY
After total gastrectomy, cobalamin deficiency is inevitable, and prophylactic cobalamin therapy should be commenced immediately after the operation. After partial gastrectomy, 10–15% of patients also develop this deficiency. The exact incidence and time of onset are most influenced by the size of the resection and the preexisting size of cobalamin body stores.
FOOD COBALAMIN MALABSORPTION
Failure of release of cobalamin from binding proteins in food is believed to be responsible for this condition, which is more common in the elderly. It is associated with low serum cobalamin levels, with or without raised serum levels of MMA and homocysteine. Typically, these patients have normal cobalamin absorption, as measured with crystalline cobalamin, but show malabsorption when a modified test using food-bound cobalamin is used. The frequency of progression to severe cobalamin deficiency and the reasons for this progression are not clear.
INTESTINAL CAUSES OF COBALAMIN MALABSORPTION
Intestinal Stagnant Loop Syndrome Malabsorption of cobalamin occurs in a variety of intestinal lesions in which there is colonization of the upper small intestine by fecal organisms. This may occur in patients with jejunal diverticulosis, enteroanastomosis, or an intestinal stricture or fistula or with an anatomic blind loop due to Crohn’s disease, tuberculosis, or an operative procedure.
Ileal Resection Removal of ≥1.2 m of terminal ileum causes malabsorption of cobalamin. In some patients after ileal resection, particularly if the ileocecal valve is incompetent, colonic bacteria may contribute further to the onset of cobalamin deficiency.
Selective Malabsorption of Cobalamin with Proteinuria (Imerslund’s Syndrome; Imerslund-Gräsbeck Syndrome; Congenital Cobalamin Malabsorption; Autosomal Recessive Megaloblastic Anemia; MGA1) This autosomally recessive disease is the most common cause of megaloblastic anemia due to cobalamin deficiency in infancy in Western countries. More than 200 cases have been reported, with familial clusters in Finland, Norway, the Middle East, and North Africa. The patients secrete normal amounts of IF and gastric acid but are unable to absorb cobalamin. In Finland, impaired synthesis, processing, or ligand binding of cubilin due to inherited mutations is found. In Norway, mutation of the gene for AMN has been reported. Other tests of intestinal absorption are normal. Over 90% of these patients show nonspecific proteinuria, but renal function is otherwise normal and renal biopsy has not shown any consistent renal defect. A few have shown aminoaciduria and congenital renal abnormalities, such as duplication of the renal pelvis.
Tropical Sprue Nearly all patients with acute and subacute tropical sprue show malabsorption of cobalamin; this may persist as the principal abnormality in the chronic form of the disease, when the patient may present with megaloblastic anemia or neuropathy due to cobalamin deficiency. Absorption of cobalamin usually improves after antibiotic therapy and, in the early stages, folic acid therapy.
Fish Tapeworm Infestation The fish tapeworm (Diphyllobothrium latum) lives in the small intestine of humans and accumulates cobalamin from food, rendering the cobalamin unavailable for absorption. Individuals acquire the worm by eating raw or partly cooked fish. Infestation is common around the lakes of Scandinavia, Germany, Japan, North America, and Russia. Megaloblastic anemia or cobalamin neuropathy occurs only in those with a heavy infestation.
Gluten-Induced Enteropathy Malabsorption of cobalamin occurs in ~30% of untreated patients (presumably those in whom the disease extends to the ileum). Cobalamin deficiency is not severe in these patients and is corrected with a gluten-free diet.
Severe Chronic Pancreatitis In this condition, lack of trypsin is thought to cause dietary cobalamin attached to gastric non-IF (R) binder to be unavailable for absorption. It also has been proposed that in pancreatitis, the concentration of calcium ions in the ileum falls below the level needed to maintain normal cobalamin absorption.
HIV Infection Serum cobalamin levels tend to fall in patients with HIV infection and are subnormal in 10–35% of those with AIDS. Malabsorption of cobalamin not corrected by IF has been shown in some, but not all, patients with subnormal serum cobalamin levels. Cobalamin deficiency sufficiently severe to cause megaloblastic anemia or neuropathy is rare.
Zollinger-Ellison Syndrome Malabsorption of cobalamin has been reported in the Zollinger-Ellison syndrome. It is thought that there is a failure to release cobalamin from R-binding protein due to inactivation of pancreatic trypsin by high acidity, as well as interference with IF binding of cobalamin.
Radiotherapy Both total-body irradiation and local radiotherapy to the ileum (e.g., as a complication of radiotherapy for carcinoma of the cervix) may cause malabsorption of cobalamin.
Graft-versus-Host Disease This commonly affects the small intestine. Malabsorption of cobalamin due to abnormal gut flora, as well as damage to ileal mucosa, is common.
Drugs The drugs that have been reported to cause malabsorption of cobalamin are listed in Table 105-4. However, megaloblastic anemia due to these drugs is rare.
ABNORMALITIES OF COBALAMIN METABOLISM
Congenital Transcobalamin II Deficiency or Abnormality Infants with TC II deficiency usually present with megaloblastic anemia within a few weeks of birth. Serum cobalamin and folate levels are normal, but the anemia responds to massive (e.g., 1 mg three times weekly) injections of cobalamin. Some cases show neurologic complications. The protein may be present but functionally inert. Genetic abnormalities found include mutations of an intra-exonic cryptic splice site, extensive deletion, single nucleotide deletion, nonsense mutation, and an RNA editing defect. Malabsorption of cobalamin occurs in all cases, and serum immunoglobulins are usually reduced. Failure to institute adequate cobalamin therapy or treatment with folic acid may lead to neurologic damage.
Congenital Methylmalonic Acidemia and Aciduria Infants with this abnormality are ill from birth with vomiting, failure to thrive, severe metabolic acidosis, ketosis, and mental retardation. Anemia, if present, is normocytic and normoblastic. The condition may be due to a functional defect in either mitochondrial methylmalonyl CoA mutase or its cofactor adocobalamin. Mutations in the methylmalonyl CoA mutase are not responsive, or only poorly responsive, to treatment with cobalamin. A proportion of infants with failure of adocobalamin synthesis respond to cobalamin in large doses. Some children have combined methylmalonic aciduria and homocystinuria due to defective formation of both cobalamin coenzymes. This usually presents in the first year of life with feeding difficulties, developmental delay, microcephaly, seizures, hypotonia, and megaloblastic anemia.
Acquired Abnormality of Cobalamin Metabolism: Nitrous Oxide Inhalation Nitrous oxide (N2O) irreversibly oxidizes methylcobalamin to an inactive precursor; this inactivates methionine synthase. Megaloblastic anemia has occurred in patients undergoing prolonged N2O anesthesia (e.g., in intensive care units). A neuropathy resembling cobalamin neuropathy has been described in dentists and anesthetists who are exposed repeatedly to N2O. Methylmalonic aciduria does not occur as adocobalamin is not inactivated by N2O.
CAUSES OF FOLATE DEFICIENCY
CAUSES OF FOLATE DEFICIENCY |
aIn severely folate-deficient patients with causes other than those listed under Dietary, poor dietary intake is often present. bDrugs inhibiting dihydrofolate reductase are discussed in the text.
NUTRITIONAL
Dietary folate deficiency is common. Indeed, in most patients with folate deficiency a nutritional element is present. Certain individuals are particularly prone to have diets containing inadequate amounts of folate (Table 128-5). In the United States and other countries where fortification of the diet with folic acid has been adopted, the prevalence of folate deficiency has dropped dramatically and is now almost restricted to high-risk groups with increased folate needs. Nutritional folate deficiency occurs in kwashiorkor and scurvy and in infants with repeated infections or those who are fed solely on goats’ milk, which has a low folate content.
MALABSORPTION
Malabsorption of dietary folate occurs in tropical sprue and in gluten-induced enteropathy. In the rare congenital recessive syndrome of selective malabsorption of folate due to mutation of the proton-coupled folate transporter (PCFT), there is an associated defect of folate transport into the cerebrospinal fluid, and these patients show megaloblastic anemia, which responds to physiologic doses of folic acid given parenterally but not orally. They also show mental retardation, convulsions, and other central nervous system abnormalities. Minor degrees of malabsorption may also occur after jejunal resection or partial gastrectomy, in Crohn’s disease, and in systemic infections, but in these conditions, if severe deficiency occurs, it is usually largely due to poor nutrition. Malabsorption of folate has been described in patients receiving sulfasalazine (Salazopyrin), cholestyramine, and triamterene.
EXCESS UTILIZATION OR LOSS
Pregnancy Folate requirements are increased by 200–300 μg to ~400 μg daily in a normal pregnancy, partly because of transfer of the vitamin to the fetus but mainly because of increased folate catabolism due to cleavage of folate coenzymes in rapidly proliferating tissues. Megaloblastic anemia due to this deficiency is prevented by prophylactic folic acid therapy. It occurred in 0.5% of pregnancies in the UK and other Western countries before prophylaxis with folic acid, but the incidence is much higher in countries where the general nutritional status is poor.
Prematurity A newborn infant, whether full term or premature, has higher serum and red cell folate concentrations than does an adult. However, a newborn infant’s demand for folate has been estimated to be up to 10 times that of adults on a weight basis, and the neonatal folate level falls rapidly to the lowest values at about 6 weeks of age. The falls are steepest and are liable to reach subnormal levels in premature babies, a number of whom develop megaloblastic anemia responsive to folic acid at about 4–6 weeks of age. This occurs particularly in the smallest babies (<1500 g birth weight) and those who have feeding difficulties or infections or have undergone multiple exchange transfusions. In these babies, prophylactic folic acid should be given.
Hematologic Disorders Folate deficiency frequently occurs in chronic hemolytic anemia, particularly in sickle cell disease, autoimmune hemolytic anemia, and congenital spherocytosis. In these and other conditions of increased cell turnover (e.g., myelofibrosis, malignancies), folate deficiency arises because it is not completely reutilized after performing coenzyme functions.
Inflammatory Conditions Chronic inflammatory diseases such as tuberculosis, rheumatoid arthritis, Crohn’s disease, psoriasis, exfoliative dermatitis, bacterial endocarditis, and chronic bacterial infections cause deficiency by reducing the appetite and increasing the demand for folate. Systemic infections also may cause malabsorption of folate. Severe deficiency is virtually confined to the patients with the most active disease and the poorest diet.
Homocystinuria This is a rare metabolic defect in the conversion of homocysteine to cystathionine. Folate deficiency occurring in most of these patients may be due to excessive utilization because of compensatory increased conversion of homocysteine to methionine.
Long-Term Dialysis Because folate is only loosely bound to plasma proteins, it is easily removed from plasma by dialysis. In patients with anorexia, vomiting, infections, and hemolysis, folate stores are particularly likely to become depleted. Routine folate prophylaxis is now given.
Congestive Heart Failure, Liver Disease Excess urinary folate losses of >100 μg per day may occur in some of these patients. The explanation appears to be release of folate from damaged liver cells.
ANTIFOLATE DRUGS
A large number of epileptics who are receiving long-term therapy with phenytoin or primidone, with or without barbiturates, develop low serum and red cell folate levels. The exact mechanism is unclear. Alcohol may also be a folate antagonist, as patients who are drinking spirits may develop megaloblastic anemia that will respond to normal quantities of dietary folate or to physiologic doses of folic acid only if alcohol is withdrawn. Macrocytosis of red cells is associated with chronic alcohol intake even when folate levels are normal. Inadequate folate intake is the major factor in the development of deficiency in spirit-drinking alcoholics. Beer is relatively folate-rich in some countries, depending on the technique used for brewing.
The drugs that inhibit DHF reductase include methotrexate, pyrimethamine, and trimethoprim. Methotrexate has the most powerful action against the human enzyme, whereas trimethoprim is most active against the bacterial enzyme and is likely to cause megaloblastic anemia only when used in conjunction with sulfamethoxazole in patients with preexisting folate or cobalamin deficiency. The activity of pyrimethamine is intermediate. The antidote to these drugs is folinic acid (5-formyl-THF).
CONGENITAL ABNORMALITIES OF FOLATE METABOLISM
Some infants with congenital defects of folate enzymes (e.g., cyclohydrolase or methionine synthase) have had megaloblastic anemia.
DIAGNOSIS OF COBALAMIN AND FOLATE DEFICIENCIES
The diagnosis of cobalamin or folate deficiency has traditionally depended on the recognition of the relevant abnormalities in the peripheral blood and analysis of the blood levels of the vitamins.
COBALAMIN DEFICIENCY
Serum Cobalamin This is measured by an automated enzyme-linked immunosorbent assay (ELISA) or competitive-binding luminescence assay (CBLA). Normal serum levels range from 118–148 pmol/L (160–200 ng/L) to ~738 pmol/L (1000 ng/L). In patients with megaloblastic anemia due to cobalamin deficiency, the level is usually <74 pmol/L (100 ng/L). In general, the more severe the deficiency, the lower is the serum cobalamin level. In patients with spinal cord damage due to the deficiency, levels are very low even in the absence of anemia. Values between 74 and 148 pmol/L (100 and 200 ng/L) are regarded as borderline. They may occur, for instance, in pregnancy, in patients with megaloblastic anemia due to folate deficiency. They may also be due to heterozygous, homozygous, or compound heterozygous mutations of the gene TCN1 that codes for haptocorrin (transcobalamin I). There is no clinical or hematologic abnormality. The serum cobalamin level is sufficiently robust, cost-effective, and most convenient to rule out cobalamin deficiency in the vast majority of patients suspected of having this problem. However, problems have arisen with commercial CBLA assays involving intrinsic factor in PA patients with intrinsic antibodies in serum. These antibodies may cause false normal serum vitamin B12 levels in up to 50% of cases tested. Where clinical indications of PA are strong, a normal serum vitamin B12 does not rule out the diagnosis. Serum MMA levels will be elevated in PA (see below).
Serum Methylmalonate and Homocysteine In patients with cobalamin deficiency sufficient to cause anemia or neuropathy, the serum MMA level is raised. Sensitive methods for measuring MMA and homocysteine in serum have been introduced and recommended for the early diagnosis of cobalamin deficiency, even in the absence of hematologic abnormalities or subnormal levels of serum cobalamin. Serum MMA levels fluctuate, however, in patients with renal failure. Mildly elevated serum MMA and/or homocysteine levels occur in up to 30% of apparently healthy volunteers, with serum cobalamin levels up to 258 pmol/L (350 ng/L) and normal serum folate levels; 15% of elderly subjects, even with cobalamin levels >258 pmol/L (>350 ng/L), have this pattern of raised metabolite levels. These findings bring into question the exact cutoff points for normal MMA and homocysteine levels. It is also unclear at present whether these mildly raised metabolite levels have clinical consequences.
Serum homocysteine is raised in both early cobalamin and folate deficiency but may be raised in other conditions, e.g., chronic renal disease, alcoholism, smoking, pyridoxine deficiency, hypothyroidism, and therapy with steroids, cyclosporine, and other drugs. Levels are also higher in serum than in plasma, in men than in premenopausal women, in women taking hormone replacement therapy or in oral contraceptive users, and in elderly persons and patients with several inborn errors of metabolism affecting enzymes in trans-sulfuration pathways of homocysteine metabolism. Thus, homocysteine levels must be carefully interpreted for diagnosis of cobalamin or folate deficiency.
Tests for the Cause of Cobalamin Deficiency Only vegans, strict vegetarians, or people living on a totally inadequate diet will become vitamin B12 deficient because of inadequate intake. Studies of cobalamin absorption once were widely used, but difficulty in obtaining radioactive cobalamin and ensuring that IF preparations are free of viruses has made these tests obsolete. Tests to diagnose PA include serum gastrin, which is raised; serum pepsinogen I, which is low in PA (90–92%) but also in other conditions; and gastric endoscopy. Tests for IF and parietal cell antibodies are also used, as well as tests for individual intestinal diseases.
FOLATE DEFICIENCY
Serum Folate This is also measured by an ELISA technique. In most laboratories, the normal range is from 11 nmol/L (2 μg/L) to ~82 nmol/L (15 μg/L). The serum folate level is low in all folate-deficient patients. It also reflects recent diet. Because of this, serum folate may be low before there is hematologic or biochemical evidence of deficiency. Serum folate rises in severe cobalamin deficiency because of the block in conversion of MTHF to THF inside cells; raised levels have also been reported in the intestinal stagnant loop syndrome due to absorption of bacterially synthesized folate.
Red Cell Folate The red cell folate assay is a valuable test of body folate stores. It is less affected than the serum assay by recent diet and traces of hemolysis. In normal adults, concentrations range from 880–3520 μmol/L (160–640 μg/L) of packed red cells. Subnormal levels occur in patients with megaloblastic anemia due to folate deficiency but also in nearly two-thirds of patients with severe cobalamin deficiency. False-normal results may occur if a folate-deficient patient has received a recent blood transfusion or if a patient has a raised reticulocyte count. Serum homocysteine assay is discussed earlier.
Tests for the Cause of Folate Deficiency The diet history is important. Tests for transglutaminase antibodies are performed to confirm or exclude celiac disease. If positive, duodenal biopsy is needed. An underlying disease causing increased folate breakdown should also be excluded.
TREATMENT COBALAMIN AND FOLATE DEFICIENCY
It is usually possible to establish which of the two deficiencies, folate or cobalamin, is the cause of the anemia and to treat only with the appropriate vitamin. In patients who enter the hospital severely ill, however, it may be necessary to treat with both vitamins in large doses once blood samples have been taken for cobalamin and folate assays and a bone marrow biopsy has been performed (if deemed necessary). Transfusion is usually unnecessary and inadvisable. If it is essential, packed red cells should be given slowly, one or two units only, with the usual treatment for heart failure if present. Potassium supplements have been recommended to obviate the danger of the hypokalemia but are not necessary. Occasionally, an excessive rise in platelets occurs after 1–2 weeks of therapy. Antiplatelet therapy, e.g., aspirin, should be considered if the platelet count rises to >800 × 109/L.
COBALAMIN DEFICIENCY
It is usually necessary to treat patients who have developed cobalamin deficiency with lifelong regular cobalamin injections. In the UK, the form used is hydroxocobalamin; in the United States, cyanocobalamin. In a few instances, the underlying cause of cobalamin deficiency can be permanently corrected, e.g., fish tapeworm, tropical sprue, or an intestinal stagnant loop that is amenable to surgery. The indications for starting cobalamin therapy are a well-documented megaloblastic anemia or other hematologic abnormalities and neuropathy due to the deficiency. Patients with borderline serum cobalamin levels but no hematologic or other abnormality may be followed to make sure that the cobalamin deficiency does not progress (see below). If malabsorption of cobalamin or rises in serum MMA levels have been demonstrated, however, these patients also should be given regular maintenance cobalamin therapy. Cobalamin should be given routinely to all patients who have had a total gastrectomy or ileal resection. Patients who have undergone gastric reduction for control of obesity or who are receiving long-term treatment with proton pump inhibitors should be screened and, if necessary, given cobalamin replacement.
Replenishment of body stores should be complete with six 1000-μg IM injections of hydroxocobalamin given at 3- to 7-day intervals. More frequent doses are usually used in patients with cobalamin neuropathy, but there is no evidence that they produce a better response. Allergic reactions are rare and may require desensitization or antihistamine or glucocorticoid cover. For maintenance therapy, 1000 μg hydroxocobalamin IM once every 3 months is satisfactory. Because of the poorer retention of cyanocobalamin, protocols generally use higher and more frequent doses, e.g., 1000 μg IM, monthly, for maintenance treatment.
Because a small fraction of cobalamin can be absorbed passively through mucous membranes even when there is complete failure of physiologic IF-dependent absorption, large daily oral doses (1000–2000 μg) of cyanocobalamin have been used in PA for replacement and maintenance of normal cobalamin status in, e.g., food malabsorption of cobalamin. Sublingual therapy has also been proposed for those in whom injections are difficult because of a bleeding tendency and who may not tolerate oral therapy. If oral therapy is used, it is important to monitor compliance, particularly with elderly, forgetful patients. This author prefers parenteral therapy for initial treatment, particularly in severe anemia or if a neuropathy is present, and for maintenance.
For treatment of patients with subnormal serum vitamin B12 (B12) levels with a normal MCV and no hypersegmentation of neutrophils, a negative IF antibody test in the absence of tests of B12 absorption is problematic. Some (perhaps 15%) cases may be due to TC I (HC) deficiency. Homocysteine and/or MMA measurements may help, but in the absence of these tests and with otherwise normal gastrointestinal function, repeat serum B12 assay after 6–12 months may help one decide whether to start cobalamin therapy.
Vitamin B12 injections are used in a wide variety of diseases, often neurologic, despite normal serum B12 and folate levels and a normal blood count and in the absence of randomized, double-blind, controlled trials. These conditions include multiple sclerosis and chronic fatigue syndrome/myalgic encephalomyelitis (ME). It seems probable that any benefit is due to the placebo effect of a usually painless, pink injection. In ME, oral B12 therapy, despite providing equally large amounts of B12, has not been beneficial, supporting the view of the effect of the injections being placebo only.
FOLATE DEFICIENCY
Oral doses of 5–15 mg folic acid daily are satisfactory, as sufficient folate is absorbed from these extremely large doses even in patients with severe malabsorption. The length of time therapy must be continued depends on the underlying disease. It is customary to continue therapy for about 4 months, when all folate-deficient red cells will have been eliminated and replaced by new folate-replete populations.
Before large doses of folic acid are given, cobalamin deficiency must be excluded and, if present, corrected; otherwise cobalamin neuropathy may develop despite a response of the anemia of cobalamin deficiency to folate therapy. Studies in the United States, however, suggest that there is no increase in the proportion of individuals with low serum cobalamin levels and no anemia since food fortification with folic acid, but it is unknown if there has been a change in incidence of cobalamin neuropathy.
Long-term folic acid therapy is required when the underlying cause of the deficiency cannot be corrected and the deficiency is likely to recur, e.g., in chronic dialysis or hemolytic anemias. It may also be necessary in gluten-induced enteropathy that does not respond to a gluten-free diet. Where mild but chronic folate deficiency occurs, it is preferable to encourage improvement in the diet after correcting the deficiency with a short course of folic acid. In any patient receiving long-term folic acid therapy, it is important to measure the serum cobalamin level at regular (e.g., once-yearly) intervals to exclude the coincidental development of cobalamin deficiency.
Folinic Acid (5-Formyl-THF) This is a stable form of fully reduced folate. It is given orally or parenterally to overcome the toxic effects of methotrexate or other DHF reductase inhibitors, e.g., trimethoprim or cotrimoxazole.
PROPHYLACTIC FOLIC ACID
Prophylactic folic acid is used in chronic dialysis patients and in parenteral feeds. Prophylactic folic acid has been used to reduce homocysteine levels to prevent cardiovascular disease and for cognitive function in the elderly, but there are no firm data to show any benefit.
Pregnancy In over 70 countries (but none in Europe), food is fortified with folic acid (in grain or flour) to reduce the risk of NTDs. Nevertheless, folic acid, 400 μg daily, should be given as a supplement before and throughout pregnancy to prevent megaloblastic anemia and reduce the incidence of NTDs, even in countries with fortification of the diet. The levels of fortification provide up to 400 μg daily on average in Chile, but in most countries, it is nearer to 200 μg, so periconceptual folic acid is still needed. Studies in early pregnancy show significant lack of compliance with the folic acid supplements, emphasizing the benefit of food fortification. Supplemental folic acid reduces the incidence of birth defects in babies born to diabetic mothers. In women who have had a previous fetus with an NTD, 5 mg daily is recommended when pregnancy is contemplated and throughout the subsequent pregnancy.
Infancy and Childhood The incidence of folate deficiency is so high in the smallest premature babies during the first 6 weeks of life that folic acid (e.g., 1 mg daily) should be given routinely to those weighing <1500 g at birth and to larger premature babies who require exchange transfusions or develop feeding difficulties, infections, or vomiting and diarrhea.
The World Health Organization currently recommends routine supplementation with iron and folic acid in children in countries where iron deficiency is common and child mortality, largely due to infectious diseases, is high. However, some studies suggest that in areas where malaria rates are high, this approach may increase the incidence of severe illness and death. Even where malaria is rare, there appears to be no survival benefit.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


