Courtesy of the CDC/Molly Kurnit, M.P. H.
“The fight against infantile paralysis is a fight to the finish, and the terms are unconditional surrender.”
—Franklin Delano Roosevelt, 32nd U.S. president and polio survivor (1944)
Viruses of the Enterovirus genus belong to one of the oldest and most diversified families of viruses, the Picornaviridae family. This family includes many important human and animal pathogens, such as polio-virus, hepatitis A virus, and foot-and-mouth-disease virus. The first enteroviruses discovered after poliovirus were the Coxsackie viruses, named according to the first geographical location of their isolation in Coxsackie, New York. Recent advances in next generation sequencing technology have opened the door to the discovery of new human enteroviruses that are abundant in feces specimens. However, the presence of these viruses is frequent in asymptomatic individuals and their clinical significance remains to be fully elucidated. The focus of this chapter is on poliovirus, the prototype of the genus Enterovirus, and a few nonpolio enteroviruses of clinical significance.
8.1 Brief Overview of Enteroviruses
Enteroviruses are naked viruses that contain a single-stranded positive (+) RNA genome packaged within a small (~30-nm) icosahedral-shaped capsid. Enteroviruses multiply in the mucosa of the gut. The prefix entero- comes from the Latin word meaning “intestine.” These ubiquitous pathogens are transmitted from person to person by an oral–fecal route. In the United States, it is estimated that enteroviruses cause between 5 and 10 million symptomatic infections, resulting in 30,000–50,000 hospitalizations annually. Most infections occur during childhood and usually produce lifelong immunity.
At least 70 distinct types of human enteroviruses have been identified, and over 20 recognizable infectious diseases have been determined to be associated with enterovirus infections. TABLE 8-1 provides examples of the clinical illnesses and common names of the enteroviruses with which they are associated. In general, most enterovirus infections cause a mild, self-limiting disease; however, sometimes they are associated with severe and fatal illnesses such as meningitis, encephalitis, myocarditis, and poliomyelitis. They may also trigger asthma exacerbations. When serious conditions such as meningitis and encephalitis do occur, it is the result of a spillover viremia (virus in the bloodstream that has access to other parts of the body), leading to secondary infections of nongastrointestinal cells. For example, poliomyelitis involves the destruction of spinal cord cells, and myocarditis is a condition in which the myocardial (heart) muscle is inflamed and weakened. Unlike the majority of infectious agents that cause gastroenteritis, enteroviruses are usually shed for prolonged periods (as long as 6 weeks) in feces. Poliovirus is the most thoroughly studied enterovirus prototype by the molecular virologist, providing important insights into picornavirus biology.
Table 8-1 Human Enteroviruses and Their Clinical Syndromes
| Virus | Syndrome |
|---|---|
| Polioviruses, types 1–3 | Paralysis (complete to partial muscle weakness) Aseptic meningitis Summer “fever” illness |
| Coxsackie viruses, group A, types 1–24 | Herpangina (mouth blisters or sores) Hand, foot, and mouth disease (A10, A16) “Common cold” Paralysis Aseptic meningitis Exanthema (rash) Hepatitis Infantile diarrhea Pneumonitis (allergic alveolitis) of infants Lymphatic or nodular pharyngitis (sore throat) Acute hemorrhagic conjunctivitis (type A24 variant) |
| Coxsackie viruses, group B, types 1–6 | Pericarditis, myocarditis Paralysis (infrequent) Macular skin rash Hepatitis Summer “fever” illness Upper respiratory illness and pneumonia Aseptic meningitis Pleurodynia (pain between the ribs or in the chest wall area) Meningoencephalitis and myocarditis in children |
| Echoviruses, types 1–33 | Meningitis Paralysis Encephalitis, ataxia (lack of coordination or muscle control), or Guillain-Barré syndrome Exanthema (rash) Respiratory illness Diarrhea Pericarditis and myocarditis Liver disturbance |
| Enteroviruses, types 68–116* | Pneumonia and bronchiolitis Acute hemorrhagic conjunctivitis (type 70) Paralysis (types D68, 70, 71) Meningoencephalitis (types 70, 71) Hand, foot, and mouth disease (type 71) |
*Since 1969, new enterovirus types have been assigned type numbers rather than being classified as coxsackie viruses or echoviruses. The common names of the previously identified enteroviruses have been retained. Information from Melnick, J. L. 1996. “Enteroviruses: Polioviruses, coxsackie viruses, echoviruses, and newer enteroviruses.” In: Fields, B. N., et al., eds. 1996. Fields Virology, 3rd ed. Philadelphia: Lippincott-Raven, pp. 655–712. | |
8.2 The History of Polio
The discovery of an 18th-Dynasty (1403 to 1365 BC) Egyptian stele that shows a priest with a shriveled leg characteristic of the flaccid paralysis (i.e., limp, weak, and lacking muscle tone), or “foot drop,” typical of poliomyelitis is evidence that this disease has a long history. Paralytic disease did not appear to be a significant problem until outbreaks began in northern Europe in the late 1800s, when reports of polio epidemics of notable size began to appear intermittently (TABLE 8-2). From that time on, the disease was reported seasonally (peaking in the summer months in temperate climates), and epidemics occurred more often in industrialized countries. In 1916, the United States faced one of the worst polio epidemics of the 20th century, when polio killed 6,000 people and paralyzed an additional 27,000. New York City was faced with so many cases that city officials responded by flooding the streets with 54 million gallons of water a day. They thought that better sanitation in the community would defeat polio, because increased sanitation had conquered diseases like typhoid, dysentery, and tuberculosis. Unfortunately, flooding the streets did not reduce the number of polio cases (FIGURE 8-1). Quarantine was also advised—in some cases, an entire town was quarantined—and many communities that bordered New York City forbade nonresidents from entering. By 1953, the incidence of paralytic polio in the United States was more than 20 per 100,000. Even though this rate was not as high as that of other common viral diseases, such as measles, polio became a public concern for several reasons:
Table 8-2 Timeline of the History of Poliomyelitis
| Year | Poliovirus Epidemics, Famous Cases, or Observations |
|---|---|
| 1403–1365 BC | Egyptian stele depicts man with flaccid paralysis of leg. |
| 1789 | British physician Michael Underwood describes polio. |
| 1870 | Jean-Martin Charcot performs microscopic studies and describes lesions or nerve damage in gray matter of spinal cord from polio patients. |
| 1890 | Oscar Medin describes polio outbreak in Stockholm, Sweden. |
| 1894 | First major outbreak in United States (132 cases in Vermont). |
| 1908 | K. Landsteiner and E. Popper showed that a filterable agent caused poliomyelitis in monkey animal model. |
| 1916 | Large outbreak of polio in the United States with over 9,000 cases reported in New York City alone; the disease kills 6,000 and paralyzes 27,000. |
| 1921 | FDR contracts polio and suffers severe paralysis. |
| 1928 | Philip Drinker and Louis Shaw invent the Drinker respirator (iron lung). |
| 1932 | FDR elected president of the United States. |
| 1934 | Major outbreak of polio in Los Angeles, with ~2,500 cases from May to November. |
| 1942 | The first Sister Kenny Institute opens in Minneapolis. |
| 1945 | WWII ends; many U.S. polio outbreaks result in 20,000 cases per year (1945–1949). |
| 1952 | 58,000 cases of polio in the United States. Testing of Salk killed vaccine begins. |
| 1953 | 35,000 cases of polio in the United States. |
| 1954 | Salk killed vaccine field trial sponsored by the National Foundation for Infantile Paralysis. |
| 1955 | Nationwide vaccination program begins. |
| 1958–1959 | Field trials of Sabin oral live vaccine. |
| 1961 | Sabin vaccine approved for use. |
| 1964 | Only 121 cases of polio in the United States. |
| 1979 | Multistate polio outbreaks in Amish communities in the United States. |
| 1981 | Time magazine reports post-polio syndrome among survivors of polio. |
| 1988 | WHO passes resolution to eradicate polio by the year 2000. |
| 1994 | China immunizes 80 million children against polio. |
| 1995 | India immunizes more than 87 million children against polio. |
| 1996 | CDC announces change in polio vaccination recommendations: two doses of inactivated polio vaccine (IPV) followed by two doses of oral polio vaccine (OPV). |
| 1999 | Number of world polio cases falls to ~7,000. |
| 2000 | OPV vaccination discontinued in the United States. |
| 2001 | 575 million children vaccinated in 94 countries. |
| 2005 | Polio-free areas, such as Nigeria and Sudan, experience new outbreaks. |
| 2005 | Polio outbreak in Amish community in Minnesota. |
| 2006 | Polio outbreak in Namibia (affected primarily young adults born before 1990). Namibia was declared polio-free in 2001. |
| 2010 | Highly lethal poliovirus outbreak in Congo-Brazzaville (42% fatal in males ages 15–25). |
| 2011 | Outbreak of polio war zone at Afghanistan/Pakistan border. Wild poliovirus confirmed in China. |
| 2013 | Wild poliovirus confirmed in sewage samples or cases in Egypt, Israel, Horn of Africa, West Bank and Gaza Strip, Israel, Syrian Arab Republic, and Cameroon. |
| 2014 | Southeast Asia, including India, declared polio-free; 80% of polio cases reported in Pakistan. |
| 2015 | Circulating cases of vaccine-derived poliovirus outbreaks in Myanmar, Laos, and Ukraine. |
*WHO polio outbreaks in the news reported at http://www.who.int/csr/don/archive/disease/poliomyelitis/en/. | |

FIGURE 8-1 In 1916, New York City was the epicenter of the first major polio outbreak in the United States. A New York City sanitation worker was photographed while sweeping the streets (circa 1910).
It was a disease of mysterious, seasonal appearance.
It could paralyze respiratory muscles.
It had disfiguring, crippling, and sometimes fatal effects.
In 1909, before the era of tissue culture experimentation, Karl Landsteiner and Erwin Popper reproduced poliomyelitis in two rhesus monkeys. To do this, they removed the spinal cord from a 9-year-old boy who had died from poliomyelitis and emulsified it in a salt solution. The spinal cord emulsion was injected into the peritoneal cavity of the monkeys. The monkeys became severely ill, exhibiting symptoms of poliomyelitis. Based on microscopic examination of the monkeys’ spinal cords, Landsteiner and Popper demonstrated that a “biological agent” in the emulsion had caused poliomyelitis. For the next 40 years, poliovirus research was limited because it required the use of animal models and formalin-treated central nervous system (CNS) tissue as a source of a vaccine for field trials. Appropriate tests for the safety and efficacy of the CNS tissue vaccines were lacking. Results obtained were inadequate to justify human vaccine trials. It was not until the advent of tissue culture techniques that the development of a polio vaccine became realistic.
In 1949, John Enders, Fredrick Robbins, and Thomas Weller authored a journal article in Science that described the successful cultivation of the Lansing strain of poliovirus in human embryonic, nonnervous tissue. Soon after, poliovirus was propagated in monkey kidney cells, and a number of laboratories began to work toward the development of a polio vaccine.
8.3 Clinical Features of Poliomyelitis
Poliomyelitis, or polio, is rare today because of vaccination efforts. The last cases of naturally occurring poliomyelitis (caused by wild-type polioviruses) in the United States were reported in four different states (Pennsylvania, Iowa, Wisconsin, and Missouri) in 1979. All of the cases occurred among unvaccinated Amish persons. After the 1997 transition to inactivated poliovirus vaccine (IPV) schedules for routine vaccination of children in the United States, vaccine-associated cases of poliomyelitis were dramatically reduced. In 2000 and 2009, individuals contracted the vaccine-derived poliovirus from someone who had received the live, oral poliovirus vaccine (OPV). The patients had weakened immune systems and multiple health problems.
The mouth is the portal of entry for polioviruses. Person-to-person poliovirus infections are acquired through an oral–fecal or, less frequently, oral–oral mode of transmission. Direct contact with human feces occurs with activities such as diaper changing. Indirect transmission may occur when sanitary conditions are poor, resulting in contaminated water, food, and fomites. Infants, particularly those in diapers, appear to be the most efficient transmitters of infection. The incubation period for poliomyelitis is 6–20 days but can range from 3 to 35 days. Poliovirus may be present in the human feces for 3–6 weeks.
The course of poliovirus infection is variable. Up to 95% of all poliovirus infections are unapparent or asymptomatic. Infected persons who are asymptomatic shed virus in feces and are able to transmit the virus to others. About 4–8% of poliovirus infections cause minor illness with symptoms of malaise, gastrointestinal disturbances, fever, flulike illness, and sore throat. A complete recovery occurs within a week. In 1–2% of infected persons, following a minor illness symptoms of stiffness of the neck, back, and/ or legs occur. The symptoms persist for 2–10 days followed by a complete recovery. Less than 1% of all poliovirus infections cause a major illness resulting in clinical signs involving the CNS, such as flaccid paralysis. Severe symptoms are associated with specific strains and types of poliovirus.
There are three poliovirus serotypes: 1, 2, and 3. Infection with one serotype does not confer immunity against a different serotype. Infection with serotype 1 is most likely to cause paralysis, and serotype 2 is the least likely to cause paralysis.
The term poliomyelitis is derived from the Greek words polios (“gray”), myelos (refers to “marrow”), and itis (“inflammation”). The severe form of the disease involves the inflammation and sometimes destruction of neurons located in the gray matter of the anterior horn of the spinal cord. Poliovirus infects motor neurons, not sensory neurons. Recovery can take as long as 2 years and may be incomplete; weakened muscles may be a permanent result. In paralytic poliomyelitis the inflammation is severe enough to destroy the motor nerves, and the resulting paralysis is permanent. The muscles that are commonly weakened by poliovirus infection are shown in FIGURE 8-2.
The major illness has three forms:
Spinal
Bulbar
Bulbospinal
Spinal poliomyelitis occurs when the lower spinal cord becomes infected by poliovirus, resulting in paralysis of the legs. In general, it is characterized by asymmetric paralysis (paralysis on one side of the body). Infection of the upper spinal cord and medulla by polio-virus results in bulbar poliomyelitis. Bulbar poliomyelitis causes muscle weakness, impairing a person’s ability to talk or swallow. Eventually it may cause paralysis of breathing, in which case an iron lung (FIGURE 8-3) or respirator may be required. Bulbar poliomyelitis is less common than spinal poliomyelitis. Bulbospinal poliomyelitis is a combination of bulbar and spinal paralysis.
Individuals infected by poliovirus are most infectious immediately before and 1–2 weeks after the onset of paralytic disease. About 80% of individuals who suffer from severe symptoms will experience permanent paralysis, 10% will die, and 10% will make a full recovery.
Post-Poliomyelitis Syndrome
Post-poliomyelitis syndrome (PPs) occurs in a large proportion of individuals who recover from paralytic poliomyelitis. The time between acute poliomyelitis and PPS ranges from 15 to 71 years, with an average of 36 years. Some studies suggest that individuals who contracted polio-virus after the age of 10 years and led an active, stable life free of impairment for at least 15 years are more at risk of developing PPS. Females also have a higher risk for developing PPS.
The onset of symptoms is insidious. Symptoms progress slowly, with new signs and symptoms followed by periods of stability. The following are the most common PPS symptoms:
New weakness in muscles or limbs involved at the time of acute poliomyelitis
Fatigue
Pain in the muscles and joints
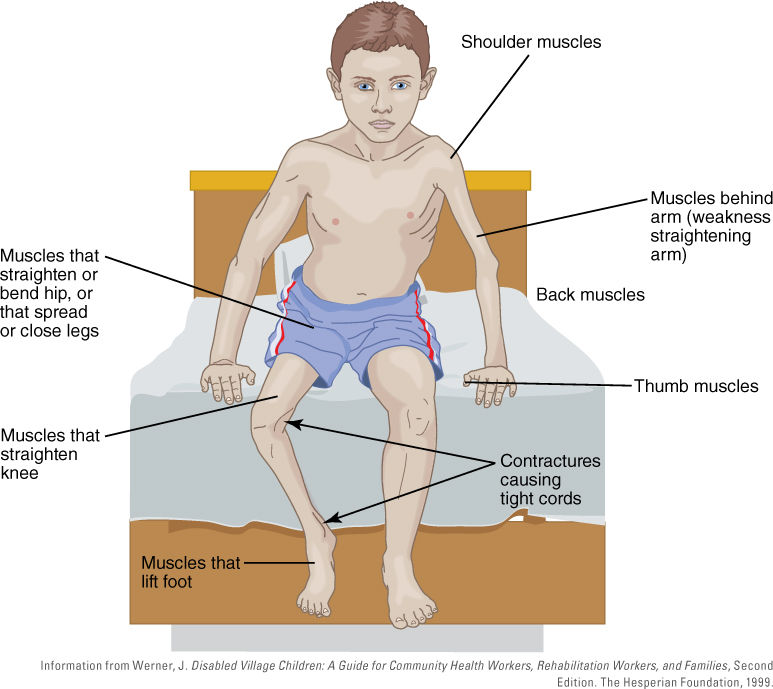
FIGURE 8-2 Muscles commonly weakened by poliomyelitis. Note that a contracture in this context is a shortening of the muscle caused by paralysis of the antagonist muscle, resulting in distortion or deformity of a body joint.

FIGURE 8-3 Mr. Barton Herbert, a polio survivor, residing in Covington, Louisiana, used this iron lung from the late 1950s until his death in 2003. Iron lungs surround the chest cavity in an airtight chamber. The chamber is used to create negative pressure around the body, causing air to rush into the lungs, aiding the patient’s breathing.
Fatigue worsens with physical activity. Other less frequent symptoms are breathing or swallowing problems, sleep-related breathing disorders such as sleep apnea, joint deformities, muscle atrophy and twitching, speech problems, and cold intolerance. PPS can result in physical disabilities and handicaps. Mobility, especially stair climbing, is most frequently affected.
The cause of PPS remains unclear. The most widely accepted hypothesis suggests that the motor neurons infected with poliovirus degenerate and die. Surviving neurons sprout new nerve endings to restore muscle function. In PPS, the muscle fibers of the surviving motor neurons slowly deteriorate over time. Eventually nerve endings are destroyed and permanent weakness results (FIGURE 8-4).

FIGURE 8-4 (a) Asymmetric atrophy of left leg in patient with PPS. (b) Clinical course of PPS. Overlapping boxes represent times when the motor neurons are becoming less stable. (c) Hypothetical model for the cause of PPS. It shows the changes in motor neurons before, during, and after poliovirus infection in individuals who experienced the paralytic form of the disease.
PPS is usually not life-threatening. Effective management of PPS requires an interdisciplinary approach using a team of physicians and healthcare personnel. The approach is based on personally tailored supportive care to manage symptoms so that the individual is comfortable and as independent as possible. Physical activity should be paced. Assistance with a cane, walker, wheel-chair, or motor scooter can help with energy conservation. Physical therapy that involves less strenuous activity to strengthen muscles, such as swimming or water aerobics, may be recommended. Occupational therapy and modification of the home environment, such as the installation of grab bars in the shower or a raised toilet seat, can improve the PPS sufferer’s ability to perform daily activities. Speech therapy can help compensate for swallowing difficulties. Sleep apnea treatments are common among PPS patients.
Over-the-counter pain medications such as aspirin, acetaminophen (Tylenol), and ibuprofen (Advil, Motrin) are recommended to ease muscle and joint pain. Experimental therapies such as the use of interferon and amantadine (antivirals), prednisone (anti-inflammatory drug), low doses of pyridostigmine (Mestinon, Regonol; nerve gas antidote), modafinil (Provigil; a drug that promotes wakefulness that is used to treat excessive daytime sleepiness associated with sleep apnea), and insulin-like growth factor (to regenerate nerve endings) have not shown any significant changes in PPS symptoms.
8.4 Classification and Structure of Poliovirus
Polioviruses are small (~30 nm in diameter), acid-stable (surviving in pH of 3.0 or lower), naked, icosahedral-shaped viruses that belong to the genus Enterovirus and family Picornaviridae (from the Greek pico, for “small”). In 1985, poliovirus serotype 1 was one of the first human viruses whose three-dimensional structure was determined in atomic detail by x-ray crystallography. The structure of the poliovirus virion consists of four distinct capsid proteins designated VP1, VP2, VP3, and VP4. The basic building block (protomer) of the capsid consists of one copy of VP1, VP2, VP3, or VP4. The capsid is made up of 60 protomers with icosahedral symmetry to form a spherical particle. The protein shell of the virus is formed by VP1, VP2, and VP3, and VP4 lies on its inner surface. VP1, VP2, and VP3 do not share amino acid sequence homology, yet they all form the same topology. As noted earlier, there are three antigenic types of poliovirus: serotypes 1, 2, and 3. All polioviruses within a serotype can be neutralized by type-specific antisera. TABLE 8-3 lists the three original poliovirus strains isolated by American scientists in the 1950s, along with the name of a strain and its origin. Today, they are known as types 1, 2, and 3. Albert Sabin attenuated three different serotypes for use as an oral live trivalent vaccine. The genome of poliovirus consists of one infectious positive-sense single-stranded RNA (+ssRNA) molecule that is 7,441 nucleotides in length. It will be described in further detail later in this chapter in the discussion of viral replication.
Stability of Enteroviruses in the Environment
Enteroviruses are able to remain infectious while passing through the stomach and the small intestine because they are unaffected by pH levels lower than 3 and are resistant to several proteases. Enterovirus particles are naked, making them resistant to detergents, 70% alcohol, and other lipid solvents, such as ether and chloroform. They are also resistant to disinfectants, such as 5% Lysol and 1% quaternary ammonium compounds (QUATS). Enteroviruses are usually present in organic matter such as feces; therefore, standard inactivation protocols may require prolonged contact time. Inactivated viruses are dead. In other words, the virus is not destroyed, but it is no longer able to infect and replicate inside of host cells. Inactivation protocols are developed to determine what procedures are necessary to decontaminate or completely disinfect surfaces where viruses are present.
Chemical inactivation of enteroviruses is achieved using chlorine, hydrochloric acid, and aldehydes. Heating at 122°F (50°C) for 1 hour in the absence of calcium and magnesium will also inactivate enteroviruses. Enteroviruses may be stable several days to several weeks at 39.2°F (4°C).
Table 8-3 Origin of the Three Original Poliovirus Strains
| Serotype | Strain | Source/Geographic Origin |
|---|---|---|
| 1 | Brunhilde | Poliovirus isolated from spinal cord of a chimpanzee, Brunhilde, who was inoculated in 1939 with stool specimens from seven paralytic patients in Baltimore, Maryland. |
| 2 | Lansing | From patient who died of paralytic polio in Lansing, Michigan, in 1938. |
| 3 | Leon | Brain and spinal cord specimen from 11-year-old boy named Leon, who died from paralytic polio in Los Angeles, California, in 1937. |
8.5 Laboratory Diagnosis of Poliovirus Infections
The United States has been free of wild poliovirus infections for more than 30 years. In a few instances, travelers contracted poliovirus through the exposure of a vaccine-derived poliovirus strain (e.g., a reverted Sabin vaccine– derived poliovirus strain). If an imported case is suspected, the state health department sends the patient’s stool sample to the CDC for laboratory testing.
Poliovirus isolation in culture is the most sensitive method to diagnose poliomyelitis. Poliovirus replicates rapidly in human and monkey cell lines (the replication cycle is complete within a few days) and is detected by visual changes in the infected cell culture, or cytopathic effects (CPEs), through microscopic observation.
Polioviruses present in patient stool samples are isolated after CPEs indicative of poliovirus infection of human or monkey kidney cells are observed. Real-time reverse transcription PCR is performed on the polioviruses isolated from the infected cell cultures. Because the nucleotide sequences of all wild-type and vaccine-type strains have been determined, real-time reverse transcription PCR is used to differentiate possible wild from vaccine-like polio-virus strains. This is called intratypic differentiation. Partial genome sequencing is used to confirm the poliovirus geno-type and determine its likely geographic origin.
Serology may be helpful in supporting the diagnosis of paralytic poliomyelitis. The serotype of the viral isolate is identified through the use of neutralization assays. With this method, antibodies are added to a virus preparation, and the infectivity of the preparation is then measured using a susceptible host system (e.g., cell cultures). A subset of antibodies produced against one serotype of poliovirus will have the ability to interfere and interact with the poliovirus and its host cell receptor. The antibodies will neutralize the infectivity of the poliovirus sero-type. Poliovirus serotype 1 is neutralized by antiserum to type 1 viruses, but not by antisera to types 2 or 3, and so on. The nucleotide sequences of all wild-type and vaccine-type strains have been determined, and as a result any poliovirus isolated from a clinical specimen collected from a person with flaccid paralysis is tested further (e.g., genomic sequencing) to determine whether the infection is caused by a vaccine-derived (e.g., a reverted Sabin vaccine–derived poliovirus strain) or wild-type virus. Improved laboratory diagnosis of poliomyelitis is an important part of the World Health Organization (WHO) initiative for global eradication of poliomyelitis.
8.6 Cellular Pathogenesis
Humans and nonhuman primates are the only natural hosts of poliovirus. Nonhuman primates have been used as models of human conditions. Mice or any other affordable experimental animals were found to be resistant to poliovirus infection, likely due to their lack of the poliovirus receptor. Natural infection occurs through the ingestion of polioviruses by an oral–fecal route of transmission.
Once ingested, polioviruses invade two lymphoid tissues: the tonsils and the Peyer’s patches (lymph nodes in the walls of the intestines near the junction of the ileum and colon). Polioviruses spread through the regional lymph nodes to the blood. A major viremia causes minor symptoms of illness in infected individuals (e.g., sore throat, headache, and fever). FIGURE 8-5 illustrates the spread of polioviruses through the bloodstream. In a very small percentage of patients, the CNS is involved. Polioviruses are carried through the bloodstream to the anterior horn cells of the spinal cord in which the virus replicates, resulting in lesions that are widely distributed throughout the spinal cord and parts of the brain. The hallmark of poliomyelitis is the selective destruction of motor neurons, which leads to paralysis and, in severe cases, respiratory arrest and death. The molecular mechanism by which poliovirus infection causes poliomyelitis is still poorly understood. This is remarkable considering that poliovirus is one of the most thoroughly investigated viruses of all time.
8.7 Poliovirus Replication
The prototype of the genus Enterovirus of the Picornaviridae family is poliovirus, the causative agent of poliomyelitis. The first step toward identifying host receptors for poliovirus came with the isolation of monoclonal antibodies that blocked binding of poliovirus to host cells. As mentioned earlier, polioviruses could not infect mouse cells because they lacked the correct cellular receptor. To bypass the receptor-binding step, the RNA genome of poliovirus was transfected into mouse cells (receptor-negative cells). It resulted in the production of a single cycle of infectious viruses. When mouse cells were transformed with human genomic DNA from HeLa cells, the mouse cells became susceptible to poliovirus infection. An antireceptor antibody blocked the poliovirus from infecting host cells. The results suggested that the poliovirus receptor gene had been introduced into mouse cells, resulting in the expression of the receptor and susceptibility to poliovirus infection.
The poliovirus receptor (PVR), (now known as CD155) is a cellular transmembrane glycoprotein and a member of the immunoglobulin (Ig) superfamily of proteins. The function of CD155 is to stimulate natural killer (NK) cells. It is now accepted that other enteroviruses may have more than one receptor, potentially expanding their host range. For example, enterovirus 71, which is frequently associated with hand, foot, and mouth disease in children, has at least two receptors: scavenger receptor B2 and P-selectin glycoprotein ligand-1. Other viruses, such as echovirus 7, bind decay-accelerating factor (DAF) but can also infect some host cells that do not contain DAF by alternatively binding to CD59 or β2-microglobulin for cellular attachment and entry.

FIGURE 8-5 Poliovirus pathogenesis.
After poliovirus attaches to its receptor, the viral capsid enters the host cell by an endocytic pathway. The viral positive-sense ssRNA, or +ssRNA, genome is released into the cytoplasm of the cell. It acts as a messenger RNA (mRNA) that is translated by ribosomes in a cap-independent manner into a single, highly autocatalytic polyprotein. The polyprotein undergoes proteolytic cleavage, resulting in capsid precursor and replication proteins.
Both ends of the poliovirus +ssRNA genome are modified. Unlike most eucaryotic mRNAs, the poliovirus genomic RNA does not contain the m7G cap structure essential for cap-dependent translation. Instead, the 5′ end of the poliovirus genome forms a cloverleaf or tRNA-like structure that plays a key role in the replicative process of the RNA molecule (FIGURE 8-6). A small peptide, VPg (“viral protein linked to the genome”) is attached to the 5′ cloverleaf end of the viral genome. VPg is essential for the initiation of viral replication. It was discovered 35 years ago and is conserved in all picornaviruses, including poliovirus, hepatitis A virus, and group A coxsackie viruses. VPgs are linked to the 5′ end of the genomic RNA. Farther downstream of the cloverleaf structure is an untranslated region (uTR) that promotes the initiation of translation by its internal ribosomal entry site (iREs). The IRES element contains extensive regions of secondary stem-loop structures. The 3′ end of the polio-virus genome is polyadenylated (Figure 8-6).

FIGURE 8-6 Schematic diagram showing the 5′ and 3′ nontranslated regions of the poliovirus genome. VPg is attached to the 5′ cloverleaf end of the viral genome. The 5′ end also contains a structural IRES element.
A poliovirus precursor polyprotein is synthesized by the translation of a single, long, open reading frame of the viral +ssRNA genome. Subsequently, the polyprotein undergoes a series of cleavages. Viral proteases cleave themselves (autocleavage) out of the singular polyprotein and cut the polyprotein into 11 separate proteins involved in replication and packaging. The viral protease 2A cleaves the p220 subunit of the cap-binding complex (eIF-4G), making host cell mRNA unrecognizable to ribosomes. This results in the shutdown of host protein synthesis but frees the ribosomes to translate viral RNA. Viral mRNA undergoes cap-independent translation by cellular ribosomes. The 5′ UTR contains the IRES, which serves as a ribosome-docking site for the 40S ribosomal subunit. IRES elements were first discovered in poliovirus, encephalomyocarditis virus (EMCV), and other picornaviruses.
IRES-dependent translation is now known as a novel mechanism of eucaryotic cap-independent translation. An increasing number of IRES elements were discovered not only in viral genomes but also in the 5′ UTRs of many cellular mRNAs. FIGURE 8-7 depicts the genomic organization and proteolytic processing of the poliovirus polyprotein. The single open reading frame is translated into a polyprotein of 247 kilodaltons (kDa) that is processed into three precursor proteins by virally encoded proteases named 2A, 3C, and 3D. The precursor proteins are further processed by virally encoded proteases (Figure 8-7). The cleavage results in four structural and seven nonstructural proteins.

FIGURE 8-7 Genomic organization and proteolytic processing of poliovirus polyprotein precursor. The viral RNA contains a VPg protein that is covalently linked to its 5′ end. The 3′ end of the viral RNA genome contains a poly (A) tail or stretch of multiple adenosine monophosphates. The genomic poliovirus RNA acts like an mRNA that is translated by host ribosomal machinery into an autocatalytic 247-kD polyprotein by the cellular ribosomal machinery in a cap-independent manner that involves the IRES. The 247-kD polyprotein is cleaved into three precursor proteins by virally encoded 2A and 3C/3CD proteases. Viral proteases continue to process the viral polyprotein. The blue circles represent poliovirus 2A, and the triangles represent poliovirus 3C/3CD proteases. The diamond shape represents an unidentified protease or an autocleavage event that occurs to process VP0. VP0 cleavage is required for the virus to be infectious; it is referred to as a maturation cleavage.
RNA replication occurs entirely in the cytoplasm of the infected cell. Host cells lack the necessary cellular machinery to replicate the viral RNA genome. Poliovirus uses a viral RNA-dependent RNA polymerase that acts as a replicase to transcribe a negative (–) ssRNA strand from the packaged genomic positive (+) ssRNA template (mRNA) that was used for protein synthesis. The first round of transcription produces a single antisense (–) ssRNA molecule followed by the synthesis of (+) sense copies of the original genome that are packaged into the viral capsids prior to viral release.

FIGURE 8-8 Poliovirus attaches to the PVR (CD155) receptor present on the host cell surface (step 1) for entry through an endocytosis mechanism (step 2). The life cycle of polioviruses occurs solely in the cytoplasm of the infected host cell. After the uncoating step, the genomic RNA is translated by cellular translational machinery through a 5′ cap-independent mechanism (ribosomes bind to the IRES) into a highly autocatalytic polyprotein (step 3) that is further cleaved into precursor, replication, and structural proteins used in capsid formation (steps 4, 5, 11). The poliovirus protein products that participate in genomic RNA synthesis are transported to membrane vesicles, the location inside of the host cell’s cytoplasm in which RNA synthesis occurs (steps 6–10). VPg is uridylated by host machinery (step 7), and subsequently acts as a protein primer for RNA replication. RNA replication involves the replication of a –ssRNA using the genomic (+) antisense ssRNA from the uncoated capsid as a template. During this process, a dsRNA replicative intermediate is partially hybridized or formed (step 8). The –ssRNA acts as a template for the synthesis of genomic (+) RNAs that are assembled into new virus particles (step 12) and released by lysis.
After the viral mRNA has been translated to produce the necessary proteins and its genome is replicated, the capsid proteins self-assemble into a structure of icosahedral symmetry that contains 60 copies of each viral capsid protein (VP1–VP4). The newly synthesized +ssRNA genome enters the incomplete capsid and is secured inside once the viral proteases finish their cleavage activity. When the genome is secured and the virion is mature, as many as 100,000 virions may be released during cell lysis of a single infected host cell. FIGURE 8-8 is an illustration of the poliovirus life or replication cycle. The +ssRNA genome of poliovirus is infectious. Polioviruses can be produced even if only the +ssRNA genome is transfected into tissue culture cells (see VIRUS FILE 8-1).
8.8 Treatments
During the era when polio outbreaks were common in the United States, new therapies were developed, such as the “Drinker respirators” or iron lungs (Figure 8-3). They were first used to assist the breathing of paralyzed patients in the 1930s. In 1940, Sister Elizabeth Kenny (1880–1952), a former staff nurse in the Australian army during World War I who established backyard clinics in the bushlands of Australia to treat long-term polio and cerebral palsy patients in Australia, arrived in Minneapolis, Minnesota. The term “sister” was a promotion she earned in 1917 in the Australian Army Nursing Service. She served on troopships bringing wounded soldiers home to Australia.
Sister Kenny became known as the nurse who challenged the physicians. Her proposed new theory to treat polio victims emphasized the movement and stretching of paralyzed muscles (physical therapy) rather than the immobilization or use of braces for paralyzed muscles as a treatment for those who suffered from the severe forms of polio. Her theory stressed the retraining, or “re-education,” of muscles so that they could function again. Other types of therapy used by Kenny included the use of hot packs and hot baths in conjunction with the active training of muscles (FIGURE 8-9). The Sister Kenny Institute opened in December 1942 in Minneapolis. Other Kenny rehabilitation clinics were established in Minnesota. In 2013, the Courage Center and Sister Kenny Rehabilitation Institute merged to form the Courage Kenny Rehabilitation Institute in Minneapolis. It provides rehabilitation services for patients with injuries and disabilities in communities throughout Minnesota and western Wisconsin.
There is no cure for polio. Treatment is supportive care, including physical therapy. Prevention through vaccination is the best way to combat infection. No anti-viral chemotherapeutic agents exist. The development of antivirals to inhibit polioviruses has seemed unnecessary because poliovirus is near eradication. No poliovirus antiviral is likely to profit a company. Only recently have the WHO and Centers for Disease Control and Prevention (CDC) called for the development of a safe antiviral that would be used to prevent and treat poliovirus infections. The motivating factors are live vaccine–associated epidemics and the potential for posteradication outbreaks.
8.9 Prevention
Poliomyelitis was the most widely known disease of the 20th century until acquired immune deficiency syndrome (AIDS). Scientists were on a mission to learn more about the disease, gradually allowing them to develop more effective treatments, rehabilitation techniques, and prevention strategies.
Chemicals and Gamma Globulin
During the 1940s, researchers observed that if certain species of monkeys were treated in their nose with chemicals such as picric acid, sodium alum, or zinc sul-fate followed by intentional infection with poliovirus, the chemicals seemed to block poliovirus infection of the nasal mucosa. When humans were treated with the same chemicals, the results were not promising and the procedure was quickly abandoned. The use of convalescent serum to treat poliomyelitis was recommended as early as 1915. In 1952, passive immunity attempts to inject 54,772 children who were at risk of contracting polio with convalescent serum had no apparent effect on the incidence or severity of paralysis, probably because passive immunity was short-lived. Another 235,000 children were inoculated with gamma globulin the following year with the same results. The use of gamma globulin failed as a prophylactic measure to prevent poliovirus infection in humans, and the supply of serum was no longer offered.

FIGURE 8-9 Sister Kenny demonstrates therapy techniques at the Sister Kenny Institute, 1942.
Inactivated Vaccines
As soon as tissue culture techniques became available, a number of medical research laboratories began to develop a vaccine against poliovirus. Dr. Jonas Salk’s laboratory began working on a formalin-inactivated, or killed, vaccine, whereas Dr. Albert Milzer and colleagues pursued ultraviolet irradiation as a means of virus inactivation. Dr. Albert Sabin and his research team took the approach of a live, attenuated vaccine.
Milzer’s vaccine was immunogenic in humans but was never adopted for general use. Salk continued work on the formalin-inactivated vaccine that was prepared by propagating the poliovirus in monkey kidney cells. By 1953, preliminary trials of Salk’s inactivated vaccine on children and adolescent volunteers at the D. T. Watson Home for Crippled Children and the Polk School for the Retarded and Feeble-Minded were conducted. During this time, medical experimentation on impoverished or mentally challenged children was accepted as a standard practice of Western medicine. Results were favorable, and the largest clinical trial of its time was set up. A mass vaccination trial that included placebo controls was sponsored by the March of Dimes (then known as the National Foundation for Infantile Paralysis). A total of 1,829,916 children from all parts of the United States, Canada, and Finland were vaccinated (FIGURE 8-10). Results of Salk’s inactivated poliovirus vaccine (iPV) were presented by manufacturers in 1955. The vaccine was said to be safe and 70% effective (booster shots would be needed to increase its efficacy). Within days, the Salk vaccine was licensed for use.
The Cutter Incident
Not long after the Salk vaccine was licensed by the U.S. Food and Drug Administration (FDA) and readily available, new cases of paralytic poliomyelitis were associated with children who received the Salk vaccine. An epidemiological investigation revealed that all of the vaccinees were injected with vaccine produced by the same manufacturer—Cutter. Infectious polioviruses were isolated from the vaccine lots, indicating that the inactivation process was not complete. A total of 260 cases were reported; 94 infections were determined to be from the vaccination, 126 were through family contacts, and 40 were through community contacts. Of these 260 cases, 192 were paralytic. There were no deaths.

FIGURE 8-10 Jonas Salk vaccinating a boy at Arsenal Elementary School, located in Pittsburgh, Pennsylvania (February 23, 1954).
Amazingly, the “Cutter incident” did not change public confidence in the Salk vaccine. New requirements were introduced for safety testing of the vaccine. A surveillance unit was created at the CDC to maintain and scrutinize all vaccination programs.
Live, Attenuated Poliovirus Vaccines
Attenuated vaccines (as opposed to inactivated vaccines) were more appealing to medical researchers because it was assumed that an active infection came closest to reproducing the natural situation. Attenuated vaccines are live, weakened forms of an infectious agent that do not cause disease but evoke an immune response after vaccination. The idea is to fool the body’s immune defense system into thinking it is under attack by a pathogenic form of the virus, thus causing it to produce the defense mechanisms (e.g., antibodies and cell-mediated responses) that will fight off the poliovirus if it is encountered in the future. It was expected that attenuated viruses would produce longer-lasting immunity. The major concern related to the creation of attenuated virus vaccines was the issue regarding the genetic stability of the poliovirus vaccine strains. In other words, would they revert to become pathogenic and cause neurovirulence after multiplying in the human host?
Three research groups worked to develop a live, attenuated poliovirus vaccine during the early 1950s. Sabin’s group obtained three different poliovirus strains and passaged (cultivated many generations in tissue culture cells or a live susceptible host) them in a variety of ways:
Polioviruses were grown in monkey testicular tissue culture cells.
Polioviruses were inoculated into the cerebrum of a rhesus monkey (a susceptible host), followed by the recovery of polioviruses excreted in the monkey’s feces. The progeny viruses from the infected monkey were used to inoculate the cerebrum of a second monkey, and the procedure was repeated in order to attenuate the polioviruses.
Chimpanzees were given food contaminated with polioviruses. The excreted strains were collected.
Chimpanzees were given food contaminated with polioviruses. The excreted strains from feces were collected and passaged in cynomolgus monkey kidney tissue culture (MKTC) cells.
Attenuation or passage histories of the three Sabin serotypes of attenuated polioviruses—called “seeds” in the vaccine production industry—are listed in TABLES 8-4, 8-5, and 8-6. FIGURE 8-11 is a schematic illustrating vaccine development by Sabin’s team. The definitive test used to determine whether the attenuated polioviruses would revert to neurovirulence (causing paralysis) involved inoculating the attenuated polioviruses intracerebrally or directly into the spinal cord of monkeys. Attenuated poliovirus serotypes or strains that passed the definitive test were used as vaccine candidates for clinical trials.
It was difficult to conduct clinical trials in the United States because the Salk vaccine had already been licensed and was used widely. In 1958, the first large-scale field trial, in which 200,000 children were vaccinated with the attenuated Sabin serotype-2 poliovirus-derived vaccine, was conducted in Singapore. The same year, laboratory protocols to cultivate the attenuated poliovirus strains in the laboratory and produce enough vaccine for 300,000 children was provided to Professor Mikhail Petrovich Chumakov, the director of the Poliomyelitis Research Institute in Moscow. Chumakov quickly manufactured more vaccine using the seed strains provided by Sabin. In just over a year, 15 million Russians were vaccinated without any untoward effects and with evidence of effectiveness. By 1960, 100 million participants in Russia and eastern European countries were vaccinated. This provided confidence that the attenuated polioviruses would not revert to being neurovirulent. After these large, successful field trials, the United States granted licensure of the Sabin live, attenuated poliovirus vaccine.
Table 8-4 Poliovirus Serotype 1, Sabin Strain, Passage History
| Year | Manipulation |
|---|---|
| 1941 | Francis and Mack: Isolation of Mahoney strain from pooled feces of three healthy children in Cleveland, Ohio. Salk: 14 rhesus monkey passages and 2 in vitro testicular tissue culture passages. |
| 1953 | Li and Schaefer: 11 monkey kidney tissue culture (MKTC) passages. Additional tissue culture passages in monkey kidney and skin. |
| 1954 | Sabin: Five passages in cynomolgus MKTC. Three single-plaque passages. Selection by neurovirulence testing. |
| 1956 | Sabin: Two passages in cynomolgus MKTC. |
| 1956 | Merck, Sharp, and Dohme: One passage in rhesus MKTC. |
Information from Sabin, A. B., and Boulger, L. R. 1973. “History of Sabin attenuated poliovirus oral live vaccine strains.” J Biol Standard 1:115–118; and Sutter, R. W., et al. “Poliovirus vaccine-live.” In Vaccines, Fifth Edition. S. Plotkin, W. Orenstein, and P. Offit, editors. Saunders, Elsevier, 2008. Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

| |