© epa european pressphoto agency b.v./Alamy Stock Photo.
“That which does not kill you makes you stronger.”
—Viktor Frankl
Each pathogen has its own infection strategy, resulting in the development of a disease with distinct symptoms. Upon viral entry, the infected host elicits a number of responses toward infection. This chapter discusses factors that affect host resistance and host defenses, including nonspecific (innate) host defenses, such as the interferon response, and specific (adaptive) immune responses, such as antibody production and cell-mediated immunity. After you finish studying this chapter, you should be able to answer the following questions:
How are viruses initially detected by the innate immune response?
How is the specific identity of the virus and the location of the infection communicated to the adaptive immune response?
How do the molecules identified by the innate system and the adaptive system differ?
How do the viral structures identified by B lymphocytes, T helpers (TH), and cytotoxic T lymphocytes (TC) differ?
Once the viral structures are identified, how do the humoral and cell-mediated arms of the adaptive immune system response act to clear infection? How do these actions actually function to clear viral infection?
What is the nature of “memory,” which all vaccines seek to produce?
5.1 Physiological Factors and Barriers Affecting Resistance
The disease triangle model addresses the interactions among the host, agent, and environment that produce disease (FIGURE 5-1). Examples of factors that contribute to disease are listed in TABLE 5-1. The disease triangle model dictates that the condition of the host impacts its susceptibility to viral infection. Prevention efforts can be directed toward any or all of the three different elements depending on the infectious disease in question and what type of intervention is available and appropriate. Several physiological factors and barriers affect resistance to viral infection. These factors include but are not limited to:
Age
Nutrition
Hormones
Fever
Genetic factors
Dual infections
Species resistance
Age
Newborns and the elderly are usually the most vulnerable to viral infections, and they tend to experience the severest symptoms. Newborns are protected from common viral infections immediately after birth through antibodies passed to them from their mothers through the placenta. This is called passive immunity. Breastfeeding also protects against infections through passive immunity from the mother during and after lactation. The “loaned” antibodies from the mother decline as the newborn begins to develop its own immune system. During this period, viral infections are frequently seen in infants. Elderly individuals become vulnerable to infection as their immune systems decline with age. There are some exceptions to this generality. Herpesvirus infections are always more severe in adults than in the young. Another exception of vulnerability to viral infection occurred during the 1918 Spanish influenza pandemic. In 1918, influenza A viruses caused severe symptoms in individuals during the prime of their lives (18–40 years) with developed and fully functioning immune systems.

FIGURE 5-1 Disease triangle model of disease causation. When these three elements interact, disease will occur. Eliminating just one of them will keep the host healthy.
Table 5-1 Disease Triangle Model: Factors Associated with Viral Infection(s)
| Causal Factors | |
|---|---|
| Susceptible host | Age, sex, nutritional status Preexisting immunity Immune suppression Genetic factors Coinfection Behavior |
| Favorable environment | Human population density (e.g., housing, crowding) Source of human exposure (e.g., housing, geography, food) Immunity of immediate contacts Incidence of infection in contacts Resistance to interventions Climate and environmental change (weather) Geography Occupational setting Air quality Food Vector/reservoir Improved water supply Sanitation |
| Viral agent (pathogen) | Survival in the environment Mode of transmission Ability to attach, invade, and multiply in the host (i.e., virulence and pathogenicity) Immunogenicity Duration of infectivity Evasion of host immunity Resistance to antiviral therapy |
Shingles, also referred to as herpes zoster, is usually a disease of the elderly or immunosuppressed (e.g., organ transplant recipients). It is caused by varicella zoster virus, the same herpesvirus that causes chickenpox or varicella. After someone is infected with varicella zoster virus, the viral DNA remains as a provirus inserted within the DNA of nuclei present in the sensory ganglionic neurons along the entire axis of the central nervous system (neuraxis). The provirus replicates silently along with the ganglia DNA and the herpesvirus infection is maintained in a dormant or latent state in which viral components are not produced and assembled to form new infectious viruses. Varicella zoster virus becomes reactivated from its latent state when the immune system is weakened due to aging, severe illness, or long-term use of corticosteroids. When the immune system cannot suppress the dormant herpesvirus any longer, the varicella zoster virus undergoes productive replication cycle. New virus components are produced and assembled into infectious virus particles in neural cell bodies that are transported to the skin, causing a painful rash that occurs along the nerve.
Shingles begins with symptoms of fatigue, chills, and fever. On the third or fourth day, the individual begins to experience a bruised feeling on one side of his or her face or body, and the affected skin area becomes excessively sensitive. Pain, tingling, itching, and prick-ling sensations begin to affect the skin, and an inflamed rash appears. The rash consists of small blisters along the path of a nerve, providing a visual symptom that is a critical part of the diagnosis (FIGURE 5-2). Shingles affects nearly 1 million people in the United States every year.
The rash is usually located on one side of the body and rarely affects the lower part of the body or the face. The blisters usually crust and heal after 5 days. Half of shingles patients, however, experience pain—often quite severe—that persists for months and sometimes years. This is called postherpetic neuralgia. Overall health and nutrition often determine the severity of the illness and length of recovery.
Nutrition
Nutritional deficiencies and malnutrition interfere with the integrity of skin and mucous membranes. This can result in an increase of infectious disease incidence in a population and in the severity of disease in an infected individual. Blood, mucus, milk, and other body fluids contain a wide variety of natural inhibitors, some of which can coat viruses and impede their attachment to cells. Therefore, any reduced activity of these natural inhibitors may increase the success of an invading virus. Besides interfering with the integrity of physical barriers and fluids that protect the host against pathogens, it has been documented that nutritional deficiencies interfere with innate immunity (e.g., functioning of complement system and phagocytes) and adaptive immunity (e.g., antibody production and cell-mediated responses). Innate and adaptive immunity are discussed in detail later in this chapter.

FIGURE 5-2 Shingles rash. Note that it follows a nerve, usually on one side of the body. Reactivation of varicella zoster virus following treatment for Hodgkin’s disease.
Besides severe malnutrition, a deficiency in essential vitamins such as vitamin A may have substantial effects on immunity. Between 100 and 140 million children in developing countries (especially those in Africa and Southeast Asia) ingest inadequate amounts of vitamin A. Children who have a vitamin A deficiency (VAD) have a 23% higher risk of dying from common infectious diseases. During a famine, the mortality rate can be as high as 50%.
VAD has a striking correlation with increased risk of common childhood diseases and even death. This is especially true for children afflicted with measles and diarrheal diseases. For this reason, one of the World Health Organization’s (WHO) goals is to eliminate VAD through interventions that include a combination of breastfeeding and vitamin A supplementation, coupled with education and promotion of vitamin A–rich diets and food fortification. In 1998, the WHO and its major partners began to deliver vitamin A supplements through immunization programs. In 2001, vitamin A was given during Polio National Immunization days in over 60 countries and was predicted to prevent over 250,000 childhood deaths. Vitamin A has been shown to act as an adjuvant to vaccines and, under certain circumstances, supplementation has been shown to enhance both cellular and humoral (antibody) responses in animals and humans. An adjuvant is an agent added to a product to enhance its effects (e.g., an adjuvant enhances the immune response stimulated by an antigen when injected with an antigen or immunogen; see the discussion in Section 5.6 on vaccine additives and side effects).
Hormones
Patients undergoing treatment with hormones such as glucocorticoids (a group of corticosteroids) are at increased risk for development of infections; for example, organ recipients undergo chronic steroid therapy. The therapy is intended to prevent rejection of the donor organ by the immune system. Common complications of this therapy include viral infections, especially cytomegalovirus and virus-related malignancies such as human herpes 8–associated Kaposi’s sarcoma.
People are continuously exposed to many different pathogens. The body produces different immune responses toward different types of pathogens. In order to gain an understanding of how an ongoing immune response toward one type of infection affects the host’s ability to respond to another pathogen, Medzhitov and colleagues used a mouse model of coinfection (see Jamieson et al., 2010, in the Resources). They discovered that influenza A virus infection in mice triggered an increase in glucocorticoid levels, which is part of a generalized stress response. In their study, the induction of glucocorticoids caused suppression of the immune response toward a subsequent bacterial infection after the mice infected with influenza A viruses were challenged with a bacterial pathogen (Listeria monocytogenes) and then studied for changes in the immune system. Influenza A virus infection is known to increase one’s susceptibility to secondary bacterial infections, particularly those opportunistic bacterial pathogens of the respiratory tract. Medzhitov’s research suggests that the mechanism of influenza A virus–mediated immune suppression is due to the increase in glucocorticoid levels. It showed that surgical removal of the adrenal glands from the mice, which are the main sources of glucocorticoids, eliminated most of the immunosuppressive effects of influenza A virus infection.
In addition to patients undergoing hormone therapies to suppress the immune system, individuals under stress may produce hormones that decrease immunity. A 2001 study published by Stowe on immune responses and short-term spaceflight concluded that astronauts experienced reactivated latent herpesvirus (Epstein-Barr virus and cytomegalovirus) infections. This reactivation was correlated with the increase of neuroendocrine hormones produced by astronauts during flight. Perhaps neuroendocrine hormones play a role in cellular immunity. Further studies are needed to determine whether immune suppression and herpesvirus reactivation will have important health consequences during long-term spaceflights. At the time of this writing, U.S. astronaut Scott Kelly and Russian Mikhail Kornienko recently returned to Earth after spending a record-breaking 340 days living in the International Space Station. Part of the space mission was to determine how space affects the immune system and more. Scientists will publish the results of their studies in the near future.
Fever
A normal, healthy individual maintains an average constant body temperature, or “set point,” of 98.6°F (37°C) that fluctuates daily about one degree above or below that number. The hypothalamus region of the brain, acting as the body’s thermostat, regulates body temperature. Fever often occurs in response to infection, inflammation, and trauma. Fever is one of the oldest clinical indicators of disease in humans and other mammalian hosts. A general definition of fever is a regulated increase in body temperature above normal daily fluctuations occurring simultaneously with an increased hypothalamic set point (above 100.5°F [38.1°C]). Evidence is mounting that fever represents complex responses of the host to various immune challenges.
Most viral infections are accompanied by a fever, or febrile response. Fever is triggered by substances called pyrogens. Pyrogens can be endogenous substances that are produced within the host or exogenous substances that are produced outside of the host. Exogenous pyrogens are essentially part or whole microorganisms, such as bacteria, parasites, or fungi, or products of microbes, such as toxins. The lipopolysaccharide (LPS) component of bacteria that possess gram-negative cell walls is the most widely studied exogenous pyrogen.
Pyrogens directly or indirectly lead to fever, and cryogens prevent excessive temperature elevation. Cryogens are host cytokines that exert antipyretic effects to protect the host from the destructive consequences of unchecked fever. Cytokines are regulatory proteins made by immune cells of the host, such as macrophages, that act on other cells to stimulate or inhibit their function. The balance of cryogens and pyrogens determines the height and duration of the fever response to any immune challenge.
Upper respiratory tract infections caused by viruses are the most common diseases of humans. Adults have 2–5 common colds each year, and children experience 7–10. A sensation of chilliness is an early symptom of common colds and is sometimes explained as an initial stage of fever because vasoconstriction of skin blood vessels (increasing blood pressure) may cause a fall in skin temperature that is perceived as chilliness.
In one study, human volunteers experienced shivering followed by chills after they were injected with an intravenous drip of typhoid vaccine (which contains known exogenous pyrogens). The volunteers, wearing swimsuits, were immersed in a water bath maintained at a neutral skin temperature of 94.1°F (34.5°C). About 65–80 minutes later, their body temperatures were rising in response to skin vasoconstriction even though they had responses of shivering and a sensation of chills. Chills and fever are likely induced by the effects of cytokines on the temperature-regulating centers of the hypothalamus (FIGURE 5-3).
Fever rarely occurs with the common cold in adults but is common in infants. This is likely the case because adults have had numerous exposures to cold viruses, and thus subsequent infections do not trigger a strong immune response. In contrast, viral infections are novel in infants, triggering a stronger immune response during their first exposures to the new viral pathogens. Fever is associated with novel or severe viral illnesses of adults infected with SARS-CoV and influenza A viruses.
Immune cells such as phagocytes produce endogenous pyrogens in response to infection. Examples of endogenous pyrogens believed to induce fever are interleukin 1 (IL-1), interleukin 6 (IL-6), interferon γ (IFN-γ), ciliary neurotrophic factor (CNTF), and tumor necrosis factor α (TNF-α). interleukins are cytokines that act to regulate the immune response, especially T lymphocyte proliferation. Cytokines are believed to cross the blood–brain barrier or interact with the vagus nerve endings to signal the hypothalamus to increase the temperature set point. The hypothalamus then initiates shivering, the sensation of chills, goose pimples, and the constriction of skin blood vessels (Figure 5-3).
Influenza viruses, myxoviruses, adenoviruses, herpesviruses, paramyxoviruses, coxsackieviruses, and Western equine encephalitis viruses induce pyrogenic effects. Others, such as vaccinia viruses and polioviruses, do not. Several vaccinia virus strains prevent fever by producing soluble IL-1 receptors. The soluble IL-1 receptors cause antipyretic effects by sequestering cellular IL-1, thereby decreasing the fever response.

FIGURE 5-3 Fever response caused by cytokines released from macrophages and other immune cells. The cytokines act on the vagal nerve endings or enter the brain to reset the temperature control center of the hypothalamus. The hypothalamus causes shivering, the sensation of chills and goose pimples, and the constriction of the blood vessels.
Genetic Factors
The outcome of a viral infection depends on the interactions between the pathogen and host. In addition to genetic variations of the viral pathogen, host genetics also play an important role in the outcome of infection. Variability in the susceptibility of humans to viral pathogens has long been observed in human history, either within a population or among different populations. When exposed to the same viral pathogen, some individuals do not appear to become infected or experience mild signs and symptoms and recover quickly after infection, whereas others succumb to the infection. Some rare individuals, about 5% of the population, appear to be resistant to human immunodeficiency virus 1 (HIV-1) infection despite repeated exposures through high-risk sexual transmission or blood transfusion. Through decades of research, it has become clear that genetic factors underlie the phenomenon of HIV resistance. Identification of such individuals has required keen observations and long-term follow-up of populations at high risk of exposure to HIV-1 infection.
Genetic polymorphisms, differences in genes themselves, involved in the replication cycle of HIV-1 infection and/or in host immune responses play an important role in the susceptibility to HIV-1 infection. For example, individuals with defective chemokine coreceptor 5 alleles (CCR5-Δ32) are largely resistant to infection by HIV-1 compared to those with functional or wild-type CCR5 alleles. HIV-positive individuals who possess an altered chemokine coreceptor 2 allele (CCR2-V64I) may progress to full-blown acquired immune deficiency syndrome (AIDS) at a much slower rate than those who do not.
HIV-1 binds to CD4, a receptor present on the outside of CD4+ TH cells, and subsequently binds to one of two coreceptors: CCR5 or CXCR4 gene products, which are chemokine coreceptors required for the entry of HIV-1 into host cells. The CCR2 gene codes for a minor alternative chemokine coreceptor used for HIV-1 entry into host cells.
The CCR5-Δ32 gene contains a premature stop codon, resulting in a truncated (shorter) chemokine coreceptor that is not present on the surface of immune cells (e.g., CD4+ T cells) of the host; in turn, HIV-1 cannot bind to the CCR5-Δ32 protein in order to enter CD4+ T cells. Individuals who are homozygous carriers for CCR5-Δ32 are largely resistant to HIV infection, whereas those who are heterozygous carriers have reduced susceptibility to HIV infection and delayed onset of AIDS. Researchers have used this knowledge to develop inhibitors that interfere with CCR5’s role in the replication cycle of HIV-1. About 12–16% of people of northern European origin and 2–5% in the Near East and India contain one or two defective CCR5-Δ32 alleles, making them largely resistant to HIV infection. In contrast, in China, where the HIV epidemic is rising, the frequency of CCR5-Δ32 alleles is absent or infrequent (FIGURE 5-4A).
The CCR2-V64I allele contains a guanine-to-adenine substitution that results in the replacement of valine with isoleucine at position 64 of the 374 amino acid CCR2 chemokine coreceptor, which has 7 transmembrane protein domains. The amino acid change of valine to isoleucine in CCR2 is associated with a 2- to 4-year delay in the progression to AIDS in HIV-infected individuals. HIV-infected individuals carrying the CCR2-V64I allele have a more favorable prognosis during antiretroviral therapy (ART).
The CCR2-V64I gene phenotype has no effect on the susceptibility to HIV-1 infection. Upon further investigation, it was discovered that the CCR2-V64I polymorphism was linked to a cytosine-to-thymine mutation in the CCR5 allele, which is located downstream of CCR2 and mapped to the human chromosome 3p21. This permutation was named CCR5-59653T. The nucleotide change may account for some effects of HIV-1 disease progression. The worldwide distribution of people carrying both the CCR2-V64I and CCR5-59653T polymorphisms has not been studied as thoroughly; however, studies to date have found that the two alleles are more common in African American and Hispanic populations than they are in Caucasian Americans (FIGURE 5-4B).

FIGURE 5-4 (a) Geographic distribution of populations containing CCR5-Δ32. The more intense the color red, the higher the allele frequency. (b) Geographic distribution of populations containing the CCR2-64K/CCR5-59653T allele. The more intense the color red, the higher the allele frequency.
The MHC/HLA (major histocompatibility complex/human leukocyte antigen) is located within a large gene region that serves as a resistance locus for nearly every human infectious disease or autoimmune disease that has been studied. This super-locus is located on human chromosome 6. It contains many genes related to immune system function. Some HIV-1 positive individuals who are elite controllers of HIV-1 infection possess polymorphisms in their HLA receptor gene located on chromosome 6 (VIRUS FILE 5-1). Elite controllers of HIV-1 infection do not progress immunologically toward AIDS. They were identified about 10 years after the HIV epidemic began as infected individuals who were able to maintain high CD4+ TH cell counts over many years despite the absence of ART therapy. Elite controllers of HIV-1 have undetectable viral loads in their blood (< 50 copies HIV-1 RNA/mL plasma), as determined by standard laboratory assays.
Several viral resistance genes have been mapped in laboratory (inbred) mice. These include the Mx (myxo-virus) and Flv (flavivirus) genes. The Mx gene product protects the mouse host during influenza A or B virus infection by interacting with the viral polymerase, thereby blocking synthesis of viral messenger RNAs (mRNAs). Later, it was discovered that the Mx gene in mice also confers resistance to other RNA viruses, such as members of the Orthomyxoviridae, Paramyxoviridae, Rhabdoviridae, Bunyaviridae, and Togaviridae families. Over the years, Mx genes homologs were discovered in the human genome. Some of these Mx proteins possess antiviral activities; others have unknown functions within the interferon system (see “The Interferon Response” in Section 5.2).
The inbred mouse Flv gene plays a role in resistance to viruses within the genus Flavivirus. This genus includes the human pathogens West Nile virus, dengue virus, and yellow fever virus. The Flv resistance gene encodes for a full-length protein within a family of proteins termed 2´5´-oligoadenylate synthetase 1b (Oas1b). Susceptible mice encode a truncated form of the protein that is missing 30% of the C-terminus because the Flv gene encodes a premature stop codon. The known function of Oas1b is to produce 2´5´-oligoadenylate polymers that, in turn, activate a latent ribonuclease, Rnase l. RNase L degrades viral and cellular single-stranded RNAs (ssRNAs). This is part of the host’s interferon response, which will be discussed in detail later in the chapter (see Section 5.2). The Oas1b proteins from both susceptible and resistant mice differ by one unique change within the protein: in the P-loop motif of the protein, a region involved in RNA recognition and binding, four amino acids are missing. It is believed that the Oas1b protein may specifically recognize and bind RNA structures unique to flavivirus RNAs.
Dual Infections
Probably the first virus that comes to mind when thinking of altered host resistance is HIV. Opportunistic infections are the hallmark of HIV infection. Patients suffering from full-blown AIDS usually die as a result of a number of secondary infections that are difficult to control. The opportunistic infections are usually caused by Candida albicans (candidiasis), Pneumocystis jiroveci (pneumonia), Toxoplasma gondii (toxoplasmosis), Cryptosporidium coccidi (cryptosporidiosis), Mycobacterium avium (pulmonary disease), Mycobacterium tuberculosis (tuberculosis), and hepatitis viruses (hepatitis). The opportunistic infections occur because of the decreased immunity caused by the depletion of TH lymphocytes (important players of the immune system) as a result of HIV-1 infection.
Viral infections of the respiratory tract such as influenza A viruses often lower the body’s resistance to secondary bacterial infections (e.g., hormonal changes, such as induction of glucocorticoids levels that cause immune suppression discussed earlier in this section), resulting in secondary pneumonia infections that can be fatal.
Species Resistance
The range of cells that can act as a host for a virus is referred to as the virus’s host range. The host range of many viruses is narrow; for example, poliovirus, HIV, and the hepatitis viruses have a very limited host range (e.g., human or other primate cells). Sometimes this is the case because host cells that are resistant to specific viral infections do not contain conserved cellular receptors located on the outside surface of cells used for viral entry, and, therefore, penetration into the cell to cause an infection cannot be achieved. Poliovirus only infects nonprimates and humans. Poliovirus binds to the receptor CD155. A study by Ida-Hosonuma and colleagues determined that poliovirus is restricted to primates because the gene sequence of the CD155 poliovirus receptor remains conserved in primates but is highly variable in other mammalian species, such as rabbits and ring-tailed lemurs, which are not susceptible to poliovirus infection. Their work has suggested that rapid changes in the CD155 gene during mammalian evolution has determined the host range restriction of polioviruses.
Besides virus–receptor interactions, viruses may be dependent on intracellular host factors for uncoating, nuclear import, and viral RNA transcription and translation of viral mRNAs. Host range is almost always determined by intracellular factors for certain DNA viruses and retroviruses. For example, simian immunodeficiency virus (SIV), a retrovirus that infects chimpanzees (SIVcpz), is thought to have crossed the species barrier into humans (to “create” HIV) because of changes in the SIVcpz matrix protein. HIV-1 does not infect rhesus macaques and a number of mammalian species because of the presence of a tripartite motif-containing protein 5 alpha, or TRIM5α. TRIM5α is a cytoplasmic protein that acts as an inhibitor, or restriction factor, of HIV-1, blocking HIV-1 infection. TRIM5α blocks uncoating by binding to the HIV-1 capsid proteins, preventing reverse transcription of the HIV-1 genome into complementary DNA (cDNA) during viral infection.
Another antiviral or restriction factor that restricts host range is an integral membrane protein named tetherin (also known as BST2, CD317, or HM1.24). It becomes incorporated into enveloped virions as they attempt to bud from infected host cells. The “captured” virions are internalized by the host cells and routed to endosomes for degradation. Ultimately, tetherin has broad antiviral activity that prevents the budding and release of enveloped viruses such as retroviruses, filoviruses, herpesviruses, and arenaviruses. Some retroviruses produce proteins that antagonize tetherin. The HIV-1 vpu and HIV-2 env gene products counter human tetherin, but the majority of SIVs use nef to counter tetherin in their simian host cells.
5.2 Host Defenses Against Viral Invaders: Nonspecific Host Defenses (Innate Immunity)
Nonspecific, or innate, immunity protects us against any pathogen, regardless of the species or type of microbe. Innate immunity is not improved by repeated exposure or contact with the pathogen. Immune cells and enzymes or proteins involved in nonspecific host defenses do not retain any “memory” from prior encounters with pathogens. Innate immunity occurs within several hours after exposure to almost any microbe or virus. It is the immunity that one is born with and serves as the first response to eliminate pathogens and prevent infection. This section will answer the following questions:
How are viruses initially detected by the innate immune response?
What kinds of viral molecules are recognized by the innate system?
Mechanical Immunity and Phagocytosis
The first line of defense against invading viruses is probably the trapping of viruses by mucus and phagocytes in the mucosal tract. This is often referred to as mechanical immunity. Phagocytosis is the engulfment and ingestion of foreign material (i.e., viruses and bacteria) by phagocytes. Examples of phagocytic cells are macrophages, neutrophils, and monocytes. Phagocytes are analogous to the video game character Pac-Man that runs through a maze while eating all of the characters in its path. Phagocytic cells patrol the tissues or blood, eating and digesting foreign invaders they encounter.
Monocytes are precursors to macrophages. They are large white blood cells containing a nucleus and granulated cytoplasm. They circulate in the bloodstream for about 8 hours, during which time they enlarge, migrate to tissues, and differentiate into macrophages. Macrophages are 5–10 times larger than monocytes and contain more lysosomes (organelles that contain many hydrolytic enzymes) than the monocytes. Some macrophages become fixed in tissues, whereas others are wandering macrophages that travel in tissues by amoeboid movement. Macrophages can ingest viruses and other foreign invaders, dead cells, cell debris, and other cellular matter. Macrophages are attracted and move toward substances using chemotactic behavior.
Macrophages patrol tissues for foreign invaders, whereas neutrophils and blood monocytes patrol circulating blood. Neutrophils are white blood cells that begin their 2-week lifespan in the bone marrow, which contains high numbers of neutrophils. This enables the body to respond to viral or other microbial challenges with a massive outpouring of neutrophils.
Some of the neutrophils migrate to the peripheral blood vessels and circulate in the blood for 7–10 hours before they migrate into tissues. Chemotactic factors released from inflamed areas attract neutrophils to the site of inflammation in the body. Neutrophils contain specialized granules that possess peroxidases, lysozymes, hydrolytic enzymes, collagenase, and lactoferrins that aid in their phagocytic activity to combat pathogens.
Viral Recognition: Pattern-Recognition Receptors (PRRs) and Pathogen-Associated Molecular Patterns (PAMPs)
The faster the host/body can detect the presence of a viral pathogen, the more quickly it can deal with it. How the body deals with viruses determines the outcome of infection. The adaptive immune system takes time to produce antibodies against viruses. Meanwhile, viruses can replicate and cause disease. Fortunately, viruses contain unique structures not present in normal tissues that can be recognized by the innate immune system.
Most body defense cells contain pattern-recognition receptors (PRRs) that detect nonself molecules, tagging them as being foreign. PRRs recognize the presence of viral pathogens through “patterns” shared by viruses called pathogen-associated molecular patterns (PAMPs). PAMPs must be recognized absolutely distinctly from natural components of cells to ensure that the host is not harmed by deleterious effects of self-recognition such as autoimmune reactions. Viral PAMPs are absent in uninfected cells and are distinct structures or molecular signatures found in viral genomes that may consist of DNA or RNA. PAMPs trigger innate immunity responses. Following the recognition of viral RNA or DNA PAMPs by host PRRs, the PRRs undergo conformational changes or specific modifications that drive cellular signaling pathways that directly inhibit viral infection.
Jules Hoffman and coworkers discovered the first PRRs in 1996 (see Lemaitre et al., 1996, in the Resources). They found that fruit flies (Drosophila sp.) with mutations in several different genes, including Toll, a gene previously demonstrated to be involved in embryonal development, could not combat microbial infections. They determined that the gene product of Toll was involved in sensing pathogenic microbes. In 1998, Bruce Beutler’s research team discovered that a gene similar to Toll in Drosophila spp. activated innate immunity against bacterial pathogens (see Poltorak et al., 1998, in the Resources). The mouse gene product responsible for the sensing was named Toll receptor. Mammals and fruit flies share similar proteins that activate innate immunity when encountering pathogenic microbes or viruses. These sensors of innate immunity were called Toll-like receptors (TLRs). TLRs are evolutionarily conserved cell-surface glycoproteins and the most thoroughly studied PRR. In 2011, the Nobel Prize in Physiology or Medicine was awarded to Bruce Beutler and Jules Hoffmann for their discoveries concerning the activation of innate immunity through TLRs.
The first PRR discovered to recognize viral nucleic acids was named Toll-like receptor 3 (TLR3). TLR3 is present on the surface of endosomes present inside of dendritic cells and macrophages and recognizes double-stranded RNA (dsRNA) viral PAMPs, triggering the induction of antiviral innate responses such as the type 1 interferon pathway (see the discussion on interferons in this section) and proinflammatory cytokines. The viral dsRNA molecules are generated as replicative intermediates during the replication cycle of most viruses. TLR7 and TLR8 recognize viral ssRNA that is rich in guanosine-uridine (GU-rich) and adenosine-uridine (AU-rich) sequences. TLR9 recognizes CpG (unmethylated cytosine-guanine oligonucleotide) PAMPs of DNA viruses present in endosomes. TABLE 5-2 contains a list of additional viral PAMPs and TLRs and the innate response triggered in dendritic cells and macrophages.
Another class of PRRs is retinoic acid-inducible gene I (RIG-I)-like receptors. RIG-I-like receptors recognize viral RNA PAMPs present in the cytoplasm of cells. FIGURE 5-5 illustrates how the diversity of viral PAMPs are detected and recognized by PRRs to induce type 1 interferons and proinflammatory cytokines.
Defensins and Viral Infection
Human α and β defensins are components of the innate immunity that have broad antibacterial and antiviral activity. Human neutrophils and epithelial cells in the gut and genitourinary tract produce α defensins. The human β defensins are produced by epithelial cells of skin and mucosal surfaces that are in contact with the environment. Enveloped and naked viruses are inhibited by the antiviral actions of α and β defensins.
Defensins are small peptides 29–42 amino acids in length that are positively charged or cationic molecules with both hydrophilic and hydrophobic (i.e., amphipathic) properties. Human cells express 6 different α defen-sins and up to 31 different β defensins. Their antiviral activity occurs through direct interactions between defensins and viruses and through indirect interactions with the infected host cell.
Table 5-2 Viral Recognition: PAMPs and PRRs in Dendritic Cells and Macrophages
| Viral PAMPs | PRR | Location | Innate Immunity |
|---|---|---|---|
| Long dsRNA | TLR3 | Endosome | Type 1 IFNs, inflammatory cytokines (e.g., TNF-α, IL-6) |
| GU-rich and AU-rich ssRNA | TLR7 | Endosome | Type 1 IFNs, inflammatory cytokines (e.g., TNF-α, IL-6) |
| GU-rich and AU-rich ssRNA | TLR8 | Endosome | Type 1 IFNs, inflammatory cytokines (e.g., TNF-α, IL-6) |
| CpG motifs of dsDNA viruses | TLR9 | Endosome | Type 1 IFNs, inflammatory cytokines (e.g., TNF-α, IL-6) |
| Viral envelope proteins | TLR2 | Cell surface | Inflammatory cytokines (e.g., TNF-α, IL-6) |
| Viral envelope proteins | TLR4 | Cell surface | Inflammatory cytokines (e.g., TNF-α, IL-6) |
| Short dsRNA or uridine-rich or adenosine-rich ssRNA | RIG-1 | Cytoplasm | Type 1 IFNs, inflammatory cytokines (e.g., TNF-α, IL-6) |
Note: TLR, Toll-like receptor; IFNs, Interferons; TNF-α, Tumor necrosis factor α; IL-6, Interleukin 6 | |||

FIGURE 5-5 Toll-like receptors and RIG-I-like receptors distinguish different viral nucleic acids and subsequently trigger or activate transcription factors such as interferon regulatory factor-3 (IRF-3), interferon regulatory factor-7 (IRF-7), or NF-κB, which turn on the expression of type 1 interferons and proinflammatory cytokines.
The α defensins have been shown to block infection of different naked viruses by such mechanisms as the aggregation or clumping of naked viruses on the surface of host cells (e.g., BK viruses are inhibited this way), the inhibition of viral uncoating (e.g., human adenoviruses), and by blocking the viral genome from entering the nucleus of its host cell (e.g., human papillomaviruses). The β defensins block enveloped viruses such as respiratory syncytial virus and human parainfluenza virus type 3 from viral entry into host cells by interacting directly with them. The defensins disrupt the viral envelope, inactivating the virus’s infectivity (i.e., ability to enter into host cells). Both α and β defensins inhibit herpes simplex virus (an enveloped virus) by blocking the cellular receptor present on the surface of its host cell, preventing entry into the host cell.
Dendritic Cells
Dendritic cells are covered with long, spiky arms that resemble the dendrites of nerve cells. Dendritic immune cells are present in tissues that have contact with the environment, such as the skin (where they are called Langerhan’s cells); mucous membranes; and the lining of the nose, lungs, and gastrointestinal tract. Medical graduate student Paul Langerhan, who was working under the mentorship of Professor Rudolf Virchow at the Institute of Pathology of the University of Berlin in 1868, first described Langerhan’s cells. In his 1868 landmark paper, “Uber die Nerven der menslichen Haut,” Langerhan described the cells as nerve endings of the skin. In 1882 he corrected his interpretation, stating, “I am now convinced that my cells are in no way essential for nerve endings.”
MHC class I and II molecules are expressed on the surfaces of dendritic cells (see Refresher: Immunology). Dendritic cells act as scouts that are very efficient at identifying foreign invaders such as viruses and bacteria, even when they are present in minute numbers (FIGURE 5-6). Dendritic cells internalize the pathogen, digest it, and display or present the foreign peptides on their surface using the MHC II molecules to TH cells. (TH cells are described later in this chapter as part of the discussion on specific immune defenses.) Activated dendritic cells also express high levels of another surface receptor, B7, which provides stimulatory signals. TH cells will only be activated to recognize the antigens as being foreign or dangerous via the B7 costimulator. After TH cells are stimulated, they leave the lymph nodes and travel to the sites of inflammation or infected tissues.

FIGURE 5-6 Colorized scanning electron micrograph of a dendritic cell. The long projections on the cell’s surface help it migrate in the upper layer of the human skin (epidermis) as it scouts for foreign antigens. Magnification: 3,500×.
Natural Killer Cells
Natural killer (NK) cells participate in early innate host defenses against viral infections and also kill tumor cells before they can establish themselves as cancers. They recognize cells that undergo a declined expression of MHC molecules or cells that contain surface antigens displayed by tumor cells or virally infected cells. NK cell deficiency in humans is associated with recurrent herpesvirus and cytomegalovirus infections, the rapid progression of cancers (especially lymphomas), hepatitis, AIDS, chronic fatigue syndrome, and various immuno-deficiency and autoimmune diseases.
In healthy people, NK cells represent 5–15% of the total lymphocyte population. They respond immediately to viral infections (within minutes to 4 hours of an infection). They are activated by the induction of IFNs α and β and other cytokines, such as IL-12, IL-15, and IL-18. The cytokines are produced by infected cells, dendritic cells, or macrophages. NK cells must be activated to do their job.
Once activated, NK cells release pore-forming proteins called perforins, granzymes (proteases), and chemokines. Perforins cause pores to form in the membranes of the target cells that are going to be killed. Granzymes activate enzyme pathways involved in apoptosis (programmed cell death). The end result is death of the target cell.
NK cells, monocytes, macrophages, and neutrophils express receptors for the fragment crystallizable (Fc) region of antibodies, as shown in FIGURE 5-7 (also see Figure 5-17). Therefore, these cells can bind to target cells containing bound antibodies. Antibodies, also referred to as immunoglobulins, are proteins typically found in blood or other bodily fluids that are part of the adaptive immune system (see Section 5.3). Antibodies identify and neutralize viruses or other foreign entities. If NK or any of the aforementioned cells interact with these target cells, they will kill them. This type of cytotoxicity is referred to as antibody-dependent cell-mediated cytotoxicity (ADCC).

FIGURE 5-7 Natural killer cells, eosinophils, macrophages, and neutrophils can bind to target (virally infected) cells containing antibodies attached to their surfaces via their Fc receptors.
The Interferon Response
Before specific immune system responses mount, the body begins both subtle and dramatic nonspecific immune responses that are induced by proteins called interferons (IFNs) at the early stage of viral infection. The ultimate outcome of this response is to stop or slow a viral infection. This is a clever way to localize infections and prevent a virus from spreading rapidly throughout the body before the immune system can finish the job. It is sometimes referred to as an infection “firebreak.”
History of IFN: The Magic Bullet
In 1957, researchers Alick Isaacs and Jean Lindenmann of the National Institute for Medical Research (NIMR) in London first described interferon in an article entitled “Virus Interference I. The Interferon,” published in the Proceedings of the Royal Society of London (see Isaacs and Lindenmann, 1957, in the Resources). Their pioneering work spurred clinical trials because interferon was speculated to be the “magic bullet” for treating viral infections, similar to what penicillin did for bacterial infections.
The Isaacs and Lindenmann experiment demonstrated that a substance “secreted by” influenza-infected cells interfered with subsequent influenza virus infections in neighboring cells (FIGURE 5-8). It was later discovered that IFNs are naturally occurring proteins that possess antiviral activity. Host cells secrete IFNs in response to viral invasion. The secreted IFNs bind to receptors on neighboring cells. This receptor-binding event acts as a signal to other cells. The end result is the creation of a cellular antiviral state, which will be discussed in greater detail later in this chapter.

FIGURE 5-8 A graphic representation of the Isaacs and Lindenmann experiment, which demonstrated that host cells produce substances that interfere with the replication of viruses.
There are a Multitude of IFNs
IFNs are cytokines—protein factors made by cells to act on other cells. In other words, they play a role in cell-to-cell communication. The number of proteins in the cytokine family continues to grow as newly discovered proteins that act on immune and nonimmune cells are added to it. Most cytokines are biochemically and structurally similar to hormones and growth factors, and they exit host cells and bind to their cell-specific cytokine receptors located on the outside of cell surfaces. This binding event activates intracellular signaling pathways, ultimately resulting in altered gene expression of target cells.
Cytokines differ from hormones and growth factors in that they are produced at very low levels within cells and are induced in response to host challenges, especially microbial infections. Here our focus is on the anti-viral activities of IFNs; however, IFNs affect a number of other processes, including the regulation of cell growth, cell differentiation, cell survival, and cell death, and play a role in infectious and inflammatory diseases, autoimmunity, and cancer.
IFN research has been complicated by the fact that there are a multitude of human IFNs, which have been classified into three types: I, II, and III. Their division is based on differences in amino acid sequence composition and the cellular receptor to which the IFN molecule binds.
Type I IFNs share amino acid sequence similarities. All type I IFNs bind to a receptor complex termed the human IFN-αβ receptor (IFN-αβR). There are at least eight different subtypes of IFN: alpha (α), beta (β), delta (δ), epsilon (∊), zeta (ζ), omega (ω), kappa (κ), and tau (τ). Type I IFNs exhibit antiviral and some antitumor activities. IFN-α has been approved for use as therapy to treat viral infections (e.g., hepatitis C and B infections) and many types of hematological cancers and solid tumors. IFN-β has been used in the treatment of multiple sclerosis. Homologs of type I IFNs are found in many species, including rats, mice, birds, reptiles, and fish; for example, IFN-τ is a unique subclass of IFNs that are transiently produced in ruminants during early pregnancy.
Humans have only one type of type II IFN: IFN-γ, which binds to the IFN-γ receptor (IFN-γR). Interferon γ is known as immune interferon, and it plays a different role, that of upregulating the immune response, compared to all other interferons. The antiviral activities of IFN-γ are very potent for some DNA viruses, including herpesviruses. However, the antitumor activities of IFN-γ are weak and have not been very useful in treating cancer.
Discovery of members of a new family of IFNs, denoted by lambda (λ), was reported in 2003. They bind to a novel receptor complex known as the class II chemokine receptor (IFN-λR). IFN-λ’s possess antiviral activity and enhance MHC I antigen expression.
The Type I IFN Pathway
This chapter focuses on the type I IFNs—IFN-α and IFN-β—because of their important roles in the induction of the antiviral state. Almost any virus-infected cell will produce IFN-α and IFN-β. The type I IFN pathway is stimulated by RIG-1-like and Toll-like receptors.
The presence of viral double-stranded RNA (dsRNA), a crucial player of the IFN pathway, triggers many of the cellular responses to virus infection. Viral dsRNA is a replication intermediate produced within cells during viral genome replication. The dsRNA must be at least 30 nucleotides in length to activate the IFN response. These long dsRNA molecules are not produced in uninfected cells. Hence, long dsRNA molecules act as a “viral infection signal” that tells the host cell to set up its antiviral state.
The long viral dsRNA molecules should not be confused with short-interfering RNAs (siRNAs). siRNAs play a role in the RNA interference (RNAi) pathway, a recently discovered gene-silencing mechanism observed in fungi, plants, invertebrates, and some vertebrates. The siRNAs are less than 25 nucleotides in length, thus escaping the IFN pathway.
dsRNA “sets up” the antiviral state by stimulating virally infected cells to produce and secrete type I IFNs, which then bind to neighboring IFN-αβRs present on the same cell or neighboring cells. The autocrine effects are quite significant during early infection.
IFNs do two key things: (1) they directly interfere with viral replication in host cells and (2) they upregu-late the expression of genes involved in the adaptive immune response toward the viral infection (e.g., genes that code for chemokines, pyrogens, MHC I molecules). The IFN receptor-binding event initiates signaling pathways that result in increased transcription of many genes. The gene products possess diverse functions, such as antiviral activity, chemokine activity, transcription regulation, and antigen presentation (discussed later in this chapter). In addition to being involved in setting up the antiviral state, IFNs halt cell proliferation of uninfected cells and alter transcription of MHC genes.
More than 20 interferon-stimulated genes (ISGs) with direct antiviral activity have been identified. More than 300 ISGs exist, but most have no known function. The list of ISGs is being compiled through the use of extensive microarray analysis of RNA samples collected from experiments on human and mice cell lines treated with IFN-α, IFN-β, or IFN-γ. An INTERFEROME open access database contains more than 28 publicly available microarray datasets (http://www.interferome.org).
Here we will discuss three well-characterized IFN-induced antiviral proteins: the dsRNA-activated protein kinase (PKR), Oas1b, and RNase L. These three proteins are normally produced in an inactive form at low levels in the cell. After IFN induction, the cell begins to express high levels of these proteins.
PKR is a serine/threonine kinase. When activated, it functions as a kinase to phosphorylate serine or threo-nine residues on itself (called autophosphorylation) or on amino acid residues of other protein targets. PKR becomes active when it binds to dsRNA, the by-product of viral replication. Once activated, PKR autophosphorylates and subsequently phosphorylates eucaryotic initiation factor 2-α (eIF-2α). eIF-2α is involved in the initiation of protein synthesis. Phosphorylation of eIF-2α blocks translation at the level of both host and viral protein synthesis. Protein synthesis is vital to the survival of cells and therefore affects viral production. PKR activity shuts down host cell activities, leading to cellular and viral incapacitation (FIGURE 5-9). Oas1b and RNase L are IFN-induced antiviral proteins that act in concert. The enzyme Oas1b is activated by viral dsRNA, converting adenine triphosphate (ATP) into 2´5´-A oligomers of adenylic acid (A). The 2´5´-A oligomers activate a latent ribonuclease, RNase L. RNase L catalyzes the degradation of both viral and host cellular RNAs, resulting in a cell death (Figure 5-9).
IFN-Resistant Viruses: The Battle Against PKR
Different viruses have evolved mechanisms to overcome the antiviral effects of PKR. Strategies that some viruses use involve the synthesis of viral products, RNA, or proteins, which bind and sequester dsRNA or bind directly to PKR, disabling inhibition of the cellular protein synthesis machinery.
Marketing IFN
The IFN story represents the complexities involved in the development of commercial drug therapies. Immediately after their discovery, IFNs were viewed as a potential antiviral panacea. However, researchers who tried to repeat Isaacs and Lindenmann’s experiments were unsuccessful. Skeptics nicknamed interferon “misinterpreton.” In spite of this, there remained a small group of believers, and the press coverage about interferon did not go unnoticed. By May 1958, patent applications were filed in the United States, Canada, and Germany.
In June 1961, volunteers of the IFN scientific committee initiated the first human experiments. Their experiments were carried out at the Common Cold Unit (CCU), a laboratory set up by a civilian Medical Research Council (MRC) at Harnham Down near Salisbury in Wiltshire (United Kingdom). The CCU was created in 1946 to perform epidemiological research on the common cold (see reviews about the CCU by David Tyrell in the Resources). David Tyrell and another committee member inoculated themselves intranasally with a small dose of monkey IFN that they had prepared themselves. A couple of days later, they inoculated themselves with a strain of coxsackie virus that was known to cause the common cold in humans 90% of the time. Months later, the coxsackie virus could be isolated from nasal washings from both researchers. Tyrell had experienced a cold, but the other committee member had not. The IFN had not caused any side effects, and the committee members agreed that small-scale human trials were doable. At this time, the United Kingdom did not have mandatory safety standards for testing drugs nor any toxicity testing or clinical trial regulations for experiments on humans.
During the late 1960s and into the early 1970s, the second and third waves of human trials were held. About this time, Matilda Krim, a cancer biologist at New York’s Memorial Sloan-Kettering Cancer Center, began searching the scientific literature for promising new developments in the area of cancer research. IFN came up in the database as showing an inhibitory effect on tumor viruses and tumors in mice and rats. Another study suggested that IFN had an inhibitory effect on tumors unrelated to virus infection.
News relating to the potential use of IFN as a new anticancer drug resulted in worldwide media hype. This was at a time during which conventional cancer therapies consisted of radiation, radical surgery, and harsh chemotherapy with severe side effects. The National Cancer Institute (NCI) was under public attack because of the low success rate associated with “battling cancer,” and there was a growing demand for effective and less toxic remedies. IFN looked promising because it was a “natural” inhibitor that reportedly did not have toxic side effects.
The early preparations of IFN used in scientific experiments and clinical studies consisted of a crude protein fraction that contained less than 10% interferon by weight. In addition to blocking antibody synthesis and other activities, the crude IFN preparations not only contained antiviral activity but also antiprotozoal, antibacterial, and antiproliferative activities. Unanticipated side effects occurred, ranging from flulike symptoms to gastrointestinal effects that included anorexia, vomiting, diarrhea, and abdominal pain. Central nervous system effects were also noted, including depression, dizziness, and altered mental states (confusion), as were cutaneous adverse reactions, such as rashes, dry skin, and alopecia (hair loss). Occasionally, some individuals experienced cardiac difficulties. It was not known whether these were caused by IFN or contaminants in the preparation.
In 1974, Hans Strander, a physician at the Karolinksa Hospital in Stockholm, Sweden, admitted to Krim that he had been treating osteosarcoma patients since 1971 with purified IFN supplied by Finnish physician Kari Cantell (FIGURE 5-10). One of Strander’s patients went into remission and the growth of tumors in others seemed to show signs of halting. Strander was optimistic about the use of IFN and stated that he did not observe toxic side effects in his patients (see mini-review by Strander, 2007, and primary literature by Muller et al., 2005, in the Resources).

FIGURE 5-9 The type 1 interferon pathway: “cellular altruism.” Infected cells secrete interferons that bind to receptors on neighboring cells. This binding event causes the cell to express several genes, including three that are involved in setting up an antiviral state: PKR, RNase L, and 2´5´-oligo (A) synthetase. The viral replication intermediate, dsRNA, activates PKR and RNase L. The end result is the destruction of viral and host mRNAs and the inhibition of protein synthesis.

FIGURE 5-10 Hans Arthur Strander worked in Kari Cantell’s research laboratory in the 1960s.
Later, Strander’s clinical study was controversial because he did not use a control group that was not treated with IFN concurrently during the investigation; however, his results and enthusiasm were enough to motivate Krim to persist in her work on IFN. Results of further clinical trials trickled in, and IFN began to look like “the miracle drug looking for a disease.” It turned out that the key was using purified IFN, but unfortunately purifying IFN turned out to be no easy task, and several years passed before the process was perfected. Success lay in the recombinant DNA technology of the early 1980s, which allowed scientists to clone several different species of the human IFN genes and produce large amounts of it in Escherichia coli. After 20 years at a snail’s pace, IFN research finally had the opportunity to quickly move forward. In addition, a number of companies were involved in altering the IFN genes to make the IFN more effective and with fewer side effects. IFN research helped genetic engineering move forward in the discipline of biotechnology.
The IFN story is one of persistence. Development and randomized controlled clinical trials required commitments from drug companies, governments, regulators, physicians, laboratory researchers, and patients and their families. Today, IFN is used to treat a diverse range of viral diseases, immune disorders, nonmalignant tumors, and cancers. TABLE 5-3 lists therapeutic uses of IFN.
Apoptosis and Viral Infection: A Double-Edged Sword
The term apoptosis has a long history in medicine, dating back to Greek physicians over 2,000 years ago. It was used in the context of “the falling off of bones” by the Greek physician Hippocrates of Cos and “the dropping of the scabs” by Galen, one of the first experimental Greek physiologists. In the 1960s, John Kerr described the morphological changes of dying rat liver cells. He hypothesized that this type of cellular death was attributed to a deliberate sequence of events and coined the term apoptosis in a landmark paper coauthored with A. H. Wyllie and A. R. Currie that was published in the British Journal of Cancer in 1972 (see Kerr and Currie, 1972, in the Resources). In the paper, the authors suggested that cell death might occur by a controlled mechanism that they called “apoptosis.” Kerr and his colleagues continued apoptosis research; however, the topic of apoptosis was largely ignored until researchers documented a correlation between apoptosis and immune system function and regulation. The immunology connection spear-headed an explosion of research. Today, over 260,400 papers have been published in the field of apoptosis, including research in premiere journals such as Science and Nature.
Apoptosis, or programmed cell death (PCD), can serve as a cellular defense mechanism to combat viral infection. It is a way to remove cells that are no longer needed (e.g., the resorption of the tadpole tail at the time of its metamorphosis or the sloughing off of the inner lining of the uterus [i.e., the endometrium] at the start of menstruation) or that are harmful to the body (e.g., virally infected cells or cancer cells).
Apoptosis is a common cellular response to a virus infection and to cancer cells. Virally infected cells usually undergo PCD internally or externally (by NK cells or TC cells), and the same is true for cancer cells in which tumor suppressor gene products activate internal PCD or NKs or TC cells extrinsically activate PCD (FIGURE 5-11). It is also possible that a virally infected cell could cause a neighboring uninfected cell to undergo PCD if it were to shut off the production of growth factors to its neighboring cell, thereby causing cell death. A virally infected cell may also secrete factors to induce a neighboring cell to activate a PCD pathway (i.e., a type I IFN pathway).
Apoptosis is triggered by events related to the disruption of the cell cycle or to virus infection. The apoptotic response involves a cascade of cellular signaling pathways. The morphological and nuclear changes associated with apoptosis are caused by the activation of a family of proteases called caspases. Caspases are usually dormant in healthy cells but in response to cell-death stimuli are converted into active enzymes that cause morphological changes associated with protease and nuclease activity (FIGURE 5-12). For example, caspase-3 causes the breakdown of several cytoskeletal proteins, cleavage of poly (ADP-ribose) polymerase, and the degradation of the inhibitor of caspase-activated DNase (ICAD). Caspase-3 cleaves ICAD, an inhibitory chaperone protein bound to CAD. In doing so, CAD is released and active. CAD is a major apoptotic nuclease that is responsible for DNA fragmentation and chromatin condensation during apoptosis, resulting in cell death. Apoptosis changes include cell shrinkage from neighboring cells, chromatin condensation to the periphery of the nucleus, nucleolus disintegration, nuclear fragmentation, membrane blebbing, and the formation of apoptotic bodies containing cytoplasm, organelles, and nuclear DNA fragments (which resolve into a characteristic DNA ladder pattern when separated by agarose gel electrophoresis). Apoptotic bodies are engulfed by macrophages.
Table 5-3 Interferon Clinical Trials
| Type of IFN | Diseases Treated |
|---|---|
| α | Chronic hepatitis B (HBV) and C (HCV) AIDS-related Kaposi’s sarcoma and HIV infection Genital papillomavirus infection, laryngeal papillomatosis Myeloproliferative disorders (cancer spreads to the blood; e.g., childhood leukemia) Multiple myeloma (cancer involving plasma cells) Thrombocythemia (blood disease characterized by the production of too many platelets, resulting in bleeding because the blood cannot clot) Non-Hodgkin’s lymphoma and Hodgkin’s disease Gastrointestinal tumors Colorectal cancer Melanoma and basal cell carcinoma Ovarian cancer Bladder cancer Kidney cancer Mesothelioma (rare form of lung cancer, often associated with asbestos exposure) Hemangiomas (reddish-colored benign tumor with many dilated blood vessels connected to it) Nonmalignant scleroderma (autoimmune disease of connective tissue that results in the buildup of scar tissue usually of the skin but can occur in organs) Multiple sclerosis |
| β | AIDS-related Kaposi’s sarcoma and HIV infection Myeloproliferative disorders (cancer spreads to the blood; e.g., childhood leukemia) Melanoma and basal cell carcinoma Bladder cancer Kidney cancer Multiple sclerosis |
| γ | Genital papillomavirus infection, laryngeal papillomatosis Melanoma and basal cell carcinoma Ovarian cancer Kidney cancer Mesothelioma (rare form of lung cancer, often associated with asbestos exposure) Nonmalignant scleroderma (autoimmune disease of connective tissue that results in the buildup of scar tissue usually of the skin but can occur in organs) Multiple sclerosis Chronic granulomatous disease (rare inherited disease of the immune system resulting in defective phagocytes), leishmaniasis (parasitic infection), mycobacterial infections (bacterial infection), osteopetrosis (rare congenital disorder in which bones are too dense), Omenn’s syndrome (inherited immune disorder), hyperimmunoglobulinemia E (disease characterized by recurrent skin and lung infections), rheumatoid arthritis, atopic dermatitis, and psoriasis |
A number of viruses can trigger apoptosis in a wide range of infections, including those of the central nervous system and heart (TABLE 5-4). Apoptosis can be detected in the brains of patients suffering from viral infections, leading to central nervous system diseases, including patients with herpes simplex virus encephalitis, HIV-1-associated dementia, and cytomegalovirus encephalitis. Also, apoptotic cardiomyocytes have been observed in diseased human hearts in patients with active viral myocarditis. Research using animal models reveals a positive correlation between apoptosis and disease severity, suggesting that apoptosis is a pathogenic mechanism in virus-induced disease. Apoptosis and mortality rates are high in mice infected with Sindbis virus and paralysis occurs in mice infected with West Nile virus. There are exceptions in which apoptosis may not be required for pathogenesis. For example, it has been shown that the brains of mice experimentally infected with a street rabies virus isolated from silver-haired bats showed little or no apoptosis even though the silver-haired bat rabies virus has been associated with most human cases of rabies in the United States. The term street virus is used to describe a virus isolated directly from an infected animal in contrast to a laboratory-attenuated virus.
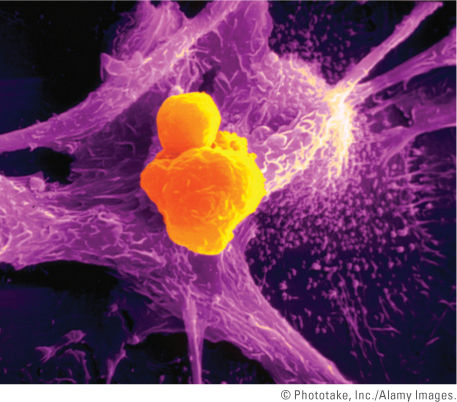
FIGURE 5-11 A colorized scanning electron micrograph that shows a TC lymphocyte (orange cell) inducing a cancer cell (pink) to undergo apoptosis. Note the pink apoptotic bodies emerging from the cancer cell. TC lymphocytes are part of the body’s immune response system. Their job is to survey, attack, and release chemicals to kill cancer cells in the body. TC lymphocytes recognize surface markers on other cells in the body that label those cells for destruction. In this way, they help to keep virus-infected or malignant cells in check.
It seems logical that viruses would be negatively affected by apoptosis. Viruses need metabolically active cells to ensure their reproduction and survival. In response to a virus infection, the host cell naturally responds by producing cytokines, including IFNs, nucleases, and proteases, all of which promote an apoptotic or cellular “suicide” event. The event localizes and prevents the spread of viruses to other cells. The host also uses immune cells such as macrophages to engulf apoptotic cells or other immune cells like T lymphocytes to induce apoptosis. It is not surprising, then, that a virus would produce an antagonist of apoptosis, but what would be the advantage of promoting apoptosis during viral infection?

FIGURE 5-12 Cellular apoptosis occurs as a result of signals that activate a cascade of proteases and nucleases that contribute to the death of the cell. Many morphological changes are associated with apoptosis, such as membrane blebbing, DNA condensation and fragmentation, and the formation of apoptotic bodies. Viruses can trigger and counteract apoptosis by mechanisms mediated by a number of different viral genes.
Table 5-4 Apoptosis Detected in Animal Models of Human Disease
| System/Organ | Virus | Animal Model | Apoptosis in Acute Disease Detected in Animal Model(s) | Human Disease |
|---|---|---|---|---|
| Central nervous system (CNS; mainly brain and spinal cord) | West Nile virus | Infects neurons of mice or golden hamsters, causing encephalitis and myelitis | Apoptosis detected in same areas as viral antigens | Fever; may progress to encephalitis, meningitis, or myelitis |
| CNS (mainly brain and spinal cord) | Sindbis virus | Infects neurons of mice, causing acute CNS disease | Apoptosis detected in neurons of brain | Causes fever, rash, meningitis, arthritis, and encephalomyelitis |
| Heart | Coxsackie virus B3 | Causes acute myocarditis associated with a low level of infected myocytes (1–3%) in some strains of mice | Apoptosis detected in cardiomyocytes | Myocarditis |
Information from Clarke, P., and Tyler, K. L. 2009. “Apoptosis in animal models of virus-induced disease.” Nat Microbiol Rev 7:144–155. Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

| ||||