
PLEOMORPHIC SOFT TISSUE TUMORS
13.1 Pleomorphic Hyalinizing Angiectatic Tumor of Soft Parts
13.3 Pleomorphic Myogenic Sarcomas (Rhabdomyosarcoma and Leiomyosarcoma)
13.5 Undifferentiated/Pleomorphic Sarcoma
13.1 Pleomorphic Hyalinizing Angiectatic Tumor of Soft Parts
Pleomorphic hyalinizing angiectatic tumor of soft parts (PHAT) is a rare lesion of soft tissue with an indeterminate lineage. So far, there are fewer than 100 cases of this neoplasm that have been reported and studied. It is considered a low-grade malignancy. It can recur and be locally aggressive, but to date, there are no reports of metastases associated with PHAT. PHAT is usually located in the subcutaneous soft tissues and has a predilection for the distal lower extremity. Occasionally it can occur more proximally, and rare cases have been described in the inguinal and axillary regions. Patients with PHAT are most commonly adults, with a median age of about 50 years. PHAT tends to be slow growing, and patients may notice evolution of the tumor over a relatively long duration (months to years). PHAT is a highly vascular neoplasm and is commonly mistaken clinically for a benign hematoma. Relatively little is known about the molecular and cytogenetic features of PHAT. Likewise, reliable information on the imaging characteristics of PHAT is sparse.
HISTOPATHOLOGY
Classic-appearing PHAT is a poorly circumscribed, spindled cell lesion with a variable amount of myxoid stroma and large ectatic vessels (Figure 13.1.1). The vessels are usually partially lined by organizing fibrin, which gives them a thick-walled appearance. There is also prominent hyalinization around the vessels in more mature lesions (Figures 13.1.2 and 13.1.3). The background stroma can be variably cellular and often focally myxoid. In early lesions (so-called “early PHAT”), there may be a diffuse proliferation of small spindled cells into subcutaneous adipose tissue, similar to the leading edge of dermatofibrosarcoma protuberans. The histologic features of early PHAT overlap substantially with those of “hemosiderotic fibrohistiocytic lipomatous lesion” (also sometimes called hemosiderotic fibrolipomatous tumor), suggesting that these may be one and the same entities. Histologic changes of “early PHAT” are often seen at the periphery of more mature lesions.
Mature or “classic” PHAT shows striking cellular atypia within the spindled component (Figures 13.1.4 and 13.1.5). This often takes the form of marked pleomorphism and hyperchromasia, as well as the presence of very prominent nuclear pseudoinclusions (Figure 13.1.6). Another frequent feature is the presence of cytoplasmic hemosiderin. An inflammatory infiltrate, largely composed of eosinophils, plasma cells, and lymphocytes, may be present as well. Despite the extensive atypia of the spindled cell component of PHAT, the mitotic activity is usually very low.
To date, cytologic features of PHAT are not well documented.
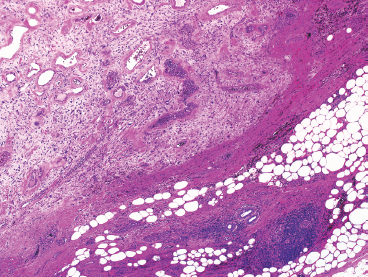
FIGURE 13.1.1 Pleomorphic hyalinizing angiectactic tumor of soft parts (PHAT) is poorly circumscribed and unencapsulated. There are often infiltrative margins.
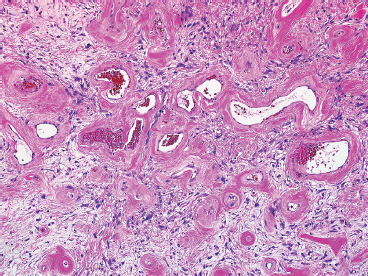
FIGURE 13.1.2 One of the most characteristic features of PHAT is the presence of numerous ectatic vessels. The stroma in the background is often very myxoid in appearance.
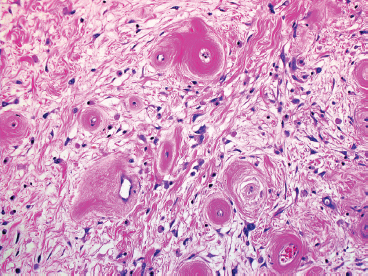
FIGURE 13.1.3 Closer inspection of more “mature” PHAT reveals vessel walls with concentric hyalinization.
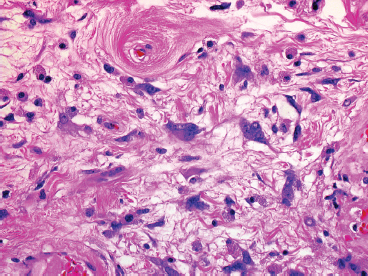
FIGURE 13.1.4 The stromal cells in PHAT tend to show marked nuclear irregularity.
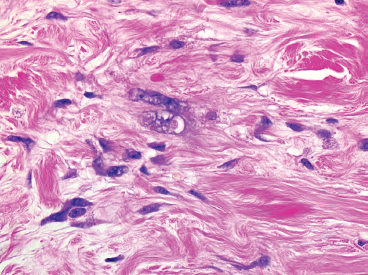
FIGURE 13.1.5 The nuclei of the stromal cells are often enlarged and have irregular configurations. Mitoses are distinctly uncommon.
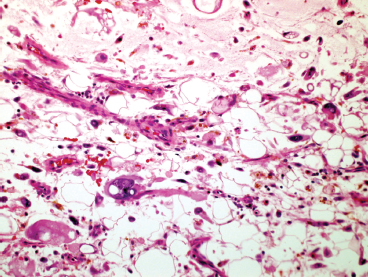
FIGURE 13.1.6 PHAT with extensive myxoid degeneration, hemosiderin-laden macrophages, and a stromal cell with a nuclear pseudoinclusion.
Pleomorphic liposarcoma (PLS) represents the rarest of the major subtypes of liposarcoma, probably accounting for about 5% of all liposarcomas. This is a high-grade, aggressive neoplasm with a tendency for multiple recurrences and metastases. PLS is largely a disease of older individuals, but rare cases in younger patients, even adolescents, have been recorded. As true of many other varieties of sarcoma, the most common site of the tumor is the deep soft tissues of the proximal extremity (Figure 13.2.1). However, this tumor has been reported in a number of unusual sites as well, including the mediastinum, head and neck region, and pericardium.
Unlike the other major subtypes of liposarcoma, PLS does not characteristically show a reproducible cytogenetic or molecular abnormality. Instead, PLS is characterized by multiple chromosome deletions, translocations, and additions. Lack of a recurrent abnormality makes the development of a “targetable” therapy for PLS problematic. Treatment is largely surgical. To date, the effects of specific chemotherapies have not been well documented.
HISTOPATHOLOGY
PLS is a high-grade neoplasm, which is most often densely cellular and very pleomorphic (Figure 13.2.2). Nonetheless, lipoblasts are easily identifiable throughout the neoplasm. They can be present in the classic univacuolated or pleomorphic multivacuolated forms (Figures 13.2.3 and 13.2.4). Necrosis and mitoses are often identified within these neoplasms. An “epithelioid” variant of PLS has been described in which the spindle cell elements are replaced either largely or completely by a more epithelioid-appearing cell. Despite the overall epithelioid look of the neoplasm, lipoblasts, as described earlier, are still a prominent feature. Rare examples of epithelioid PLS have been shown to express immunohistochemical markers of epithelial differentiation. This feature may lead to an incorrect diagnosis of sarcomatoid carcinoma. One way to avoid this mistake is to focus on the presence of the lipoblasts. These should not be present in a high-grade sarcomatous carcinoma.
CYTOLOGIC FINDINGS
On aspirates, PLS tend to resemble aspirates of renal or adrenal tumors. There is usually a dispersed fatty component in the background, which can be confused with myxoid or necrotic materials (Figure 13.2.5). One of the key features to the diagnosis is the presence of fat vacuoles within the cytoplasm of the tumor cells. These can be present as small microvesicular droplets of fat, giving a small “cookie cutter” type of vacuole or can manifest as a true lipoblast. Both types of lipoblasts described earlier, classic and pleomorphic multivacuolated, are present in aspirated materials (Figure 13.2.6).
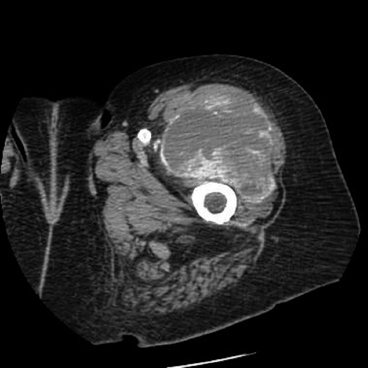
FIGURE 13.2.1 Imaging of a Pleomorphic liposarcoma (PLS) arising in the deep compartment of the proximal lower extremity.
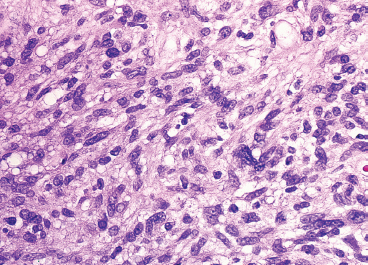
FIGURE 13.2.2 Although a highly pleomorphic tumor, fat is usually easily identified in PLS. This feature makes PLS one of the easiest of the “pleomorphic tumors” to accurately subclassify.
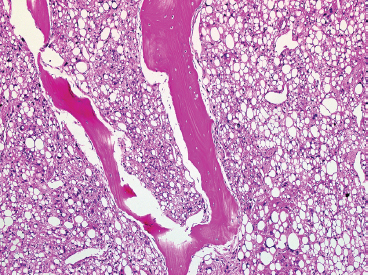
FIGURE 13.2.3 PLS metastatic to bone. PLS can easily be confused with other types of “clear cell” tumors, particularly renal cell or adrenal carcinoma.
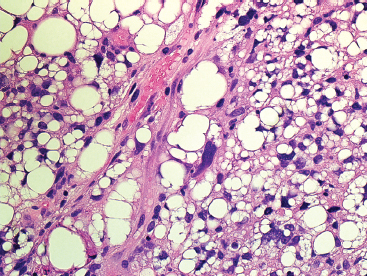
FIGURE 13.2.4 PLS typically has numerous lipoblasts, both multivacuolated and the classic single-vacuole type.
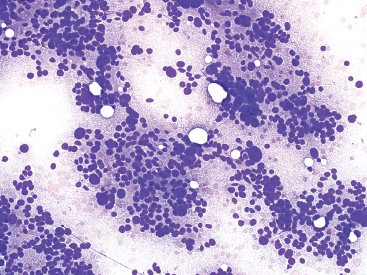
FIGURE 13.2.5 Aspirates of PLS often have a “grungy” background due to the presence of abundant extracellular lipid debris. The punched-out holes correspond to intact fat vacuoles.
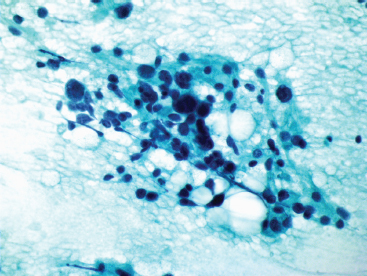
FIGURE 13.2.6 A fixed preparation from an aspirate of PLS showing cellular pleomorphism and dark nuclear chromatin.
13.3 Pleomorphic Myogenic Sarcomas (Rhabdomyosarcoma and Leiomyosarcoma)
The pleomorphic subtype of rhabdomyosarcoma is exceedingly uncommon. Unlike other types of rhabdomyosarcoma, this tumor occurs in older adults, very rarely in children. It arises most commonly in the deep soft tissues of the extremities, with rare cases in the abdomen, retroperitoneum, and head and neck regions. Patients present with a mass lesion and associated pain. Pleomorphic rhabdomyosarcoma is an aggressive neoplasm, with 80% of the patients having metastases within 5 years. Poor prognosis is associated with advanced patient age. Treatment is multimodal, with wide surgical excision and adjuvant radiation. Pleomorphic rhabdomyosarcomas are associated with complex cytogenetic abnormalities including numerical and structural changes.
Pleomorphic leiomyosarcoma represents the extreme, high-grade end of the histologic spectrum of leiomyosarcoma. Although leiomyosarcoma at any site can show pleomorphic features, the majority of leiomyosarcomas with a pleomorphic phenotype occur in the retroperitoneum or extremity. The prognosis for the pleomorphic variant of leiomyosarcoma is slightly better than that of rhabdomyosarcoma. Pleomorphic leiomyosarcoma is associated with complex cytogenetic changes.
HISTOPATHOLOGY
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


