Chapter 45 Y.M. Dennis Lo, M.A., D.M., D.Phil., F.R.C.P.(Lond. & Edin.), F.R.C.Path., F.R.S., and Rossa W.K. Chiu, M.B.B.S., Ph.D., F.H.K.A.M.(Pathology), F.R.C.P.A. It is generally agreed that cell-free nucleic acids in plasma were first discovered by Mandel and Metais in 1948.136 It was especially remarkable that this work preceded the discovery of the double-helical structure of DNA in 1953.204 However, the significance of the work by Mandel and Métais was not recognized for many years. It was not until 1966 that interest in the area re-emerged with the finding of DNA in the serum of patients with systemic lupus erythematosus.187 Similar findings in other autoimmune diseases were subsequently reported.101 In the 1970s, it was reported that increased concentrations of cell-free DNA were found in the serum of cancer patients, when compared with controls.102 Furthermore, it was shown that the reduction in concentrations of serum DNA correlated with treatment response. However, the origin of such serum DNA remained unclear. In 1989, Stroun and associates proposed that such DNA might have originated from cancer cells.182 However, definitive proof of this hypothesis had to wait until 1994, when two groups used polymerase chain reaction (PCR)-based technologies to show that oncogene mutations were indeed detectable in the plasma of patients with pancreatic cancer,179 myelodysplastic syndrome, and acute myelogenous leukemia.197 Two years later, two groups confirmed these observations by showing that tumor-derived microsatellite alterations could be found in the plasma and serum of patients with small cell lung cancer25 and head and neck cancer,146 respectively. These results laid the foundation for rapid developments in the field over the following decade (Table 45-1). TABLE 45-1 Plasma Nucleic Acid Features Associated With Tumors Several groups have replicated the previously mentioned pioneering work on plasma and serum DNA. On the simplest level, the total concentrations of DNA in plasma are measured. Thus, some groups have demonstrated that the total concentrations of DNA in the plasma of cancer patients are generally higher than those in controls.5,8,171,174,180 For example, Sozzi and colleagues showed that the median concentration of plasma DNA in lung cancer patients is almost eight times higher than that in controls.180 Similar results have been reported by Paci and coworkers.154 However, it is important to note that the total concentration of plasma/serum DNA is relatively nonspecific and could be influenced by pathologies other than cancer (e.g., tissue injury, as discussed later). Thus, most workers in the field have focused on a variety of tumor-derived genetic and other molecular alterations. Circulating DNA in plasma consists of short fragments of DNA.87 In 2003, it was shown that the DNA molecules in the plasma of gynecologic and breast cancer patients were shorter than those in the plasma of control subjects.200 In this study, the length of plasma DNA molecules was assessed by measuring the relative abundance of a longer versus a shorter fragment of DNA using quantitative PCR.200 Results were expressed as a DNA integrity index. This approach was similar to the method used to analyze the length of Epstein-Barr virus (EBV) DNA in the plasma of patients with nasopharyngeal carcinoma (NPC).20 Wang and associates attributed the success of plasma DNA size as a tumor marker to the fact that necrosis might be a relatively commonly phenomenon in tumor tissues and might be associated with the liberation of relatively longer fragments of DNA into the plasma. It was also suggested that cell death in normal tissues might be more related to apoptosis, which might be associated with the release of shorter DNA fragments into the circulation. Various groups have confirmed the potential usefulness of plasma DNA integrity analysis for cancer patients.18,73,89,196 For example, Chan and colleagues reported that the plasma DNA integrity of NPC patients was higher than that of controls.18 Furthermore, radiotherapy was associated with a reduction in the plasma DNA integrity index in 70% of patients. Most important, patients who did not demonstrate this reduction had a worse prognosis than those who had normalization in the DNA integrity index. However, Holdenrieder and coworkers and Boddy and associates were unable to reproduce the value of DNA integrity analysis for the plasma and serum of cancer patients.9,77 Chan and colleagues studied preanalytical factors associated with the measurement of DNA integrity.19 These workers showed that clotting and delayed separation of plasma from blood cells for 24 hours would significantly increase the concentration and observed size of cell-free DNA in blood samples. They also showed that repeated freezing and thawing of plasma samples, but not extracted DNA, leads to fragmentation of DNA. These factors should be taken into consideration if DNA integrity should become a clinical molecular test. Numerous groups have reported the detection of tumor-associated loss of heterozygosity (LOH) in the plasma/serum of cancer patients. As examples, this has been reported in lung cancer,68 prostate cancer,143,172 breast cancer,173 melanoma,185,186 renal cancer,69 and transitional cell carcinoma of the urinary bladder.199 Most workers have used microsatellites as the targets for such analysis. However, debate regarding the robustness of this approach is ongoing, as it has been demonstrated that PCR artifacts might occasionally result in the false identification of LOH when in fact none is present.40 It has been suggested that fractionation of plasma DNA and specific targeting of lower-molecular-weight DNA might improve the detection of LOH in plasma.143 Targeting of single-nucleotide polymorphisms (SNPs), instead of microsatellites, is an alternative method for the detection of LOH.22 Another approach for the detection of tumor-derived DNA in plasma is to target mutant gene sequences. Examples of genes that have been studied in this fashion include the KRAS oncogene in colorectal carcinoma94,166,167,183 and pancreatic carcinoma,12,58,210 the adenomatosis polyposis coli (APC) gene in colorectal carcinoma,97 and the TP53 gene (coding for the p53 protein) in lung cancer,72 ovarian cancer,152 and hepatocellular carcinoma (HCC).79,90,92 Much interest has been expressed in mutations in the epidermal growth factor receptor (EGFR) gene in non–small-cell lung cancer and its relation to response to tyrosine kinase inhibitors.133,155 It has been shown that the mutational status of the EGFR gene can be predicted from plasma/serum DNA analysis and is strongly correlated with response to tyrosine kinase inhibitor treatment.91,134,213 The main challenge faced by investigators working with the detection of tumor-derived gene mutations is that the minority mutant DNA molecules are surrounded by a “sea” of nonmutant molecules. Thus, detection methods should allow the sensitive and specific detection of such mutant sequences from the background. Many workers initially used allele-specific PCR amplification.40,91 Much promise has been shown with the use of techniques that allow analysis of single DNA molecules. One such method is single-molecule PCR or digital PCR (see Chapter 17), in which the DNA template is diluted and then amplified in a series of PCRs.198 The degree of dilution is such that each PCR will contain approximately 0.5 copy of the template. In such a situation, distribution of the template DNA molecule among the PCRs will follow the Poisson distribution,125 with some reactions containing no target molecule and other reactions containing a single target molecule. The Poisson distribution also predicts that some reactions will contain more than a single target molecule. However, the latter will be the minority at the template concentration of 0.5 copy of target per reaction. Reactions containing a single target molecule, mutant or wild-type, can be analyzed without the interference of closely related sequences. Another advantage of the digital PCR approach lies in its quantitative applications. In this regard, the number of positive reactions will reflect the number of starting template molecules. The number of reactions containing more than one template molecule can be mathematically “corrected” by using the Poisson distribution.125 The main disadvantage of digital PCR is the need to carry out numerous parallel PCRs. However, the development of several platforms for high-throughput implementation of digital PCR has offered the hope that this method might eventually be used in routine molecular diagnostics. One such method employs microfluidics to distribute the reaction mixture to hundreds or thousands of nanoliter chambers.203 This method has been used for the detection and measurement of the concentration of EGFR mutations in the plasma of non–small-cell lung cancer patients.213 Another method involves the performance of PCR in emulsions, following the capture of PCR products on beads.48 This method, which is referred to as BEAMing (beads, emulsion, amplification, and magnetics), has been used to detect and measure the concentration of mutant DNA molecules in the plasma of colorectal cancer patients.44,45 Much interest has arisen in the study of epigenetic alterations in tumors. Epigenetics is the study of molecular changes that influence gene expression, but that do not involve a change in DNA sequence.175 One of the best studied epigenetic mechanisms is DNA methylation, which typically involves the methylation of the cytosine residue of the CpG dinucleotide. DNA methylation in the promoter region of genes is associated with gene silencing.85 This is one of the mechanisms for the inactivation of tumor suppressor genes in cancer.139 In 1999, two groups first demonstrated the presence of tumor-associated DNA methylation in the plasma and serum of cancer patients.51,208 Both groups used a method known as methylation-specific PCR (MSP).75 DNA for MSP analysis is first treated with bisulfite, a process that converts unmethylated cytosine to uracil, while leaving methylated cytosine unchanged.62 Hence, methylated and unmethylated sequences can then be amplified using different primers. The versatility of MSP has since been enhanced by the development of quantitative MSP, in which the concentration of targets with different methylation status can be measured.49,117 Tumor-associated DNA methylation changes have since been detected for many tumor suppressor genes and for many types of cancer. Examples include the gene for p16 in non–small-cell lung cancer,51 colorectal cancer,145 and HCC208 patients; the SEPT9 gene (septin 9) in colorectal cancer71; the APC and several other genes in breast cancer78; and the GSTP1 gene in prostate cancer.4 The main advantage of the methylation-based approach is that one methylation assay can typically be used for several tumor types. This is different from the previously mentioned approach of targeting tumor-specific gene mutations in which multiple assays are often needed to target the many possible mutations of a gene. The main disadvantage of the MSP-based approach is that the bisulfite conversion step is known to degrade the input of DNA significantly, with certain workers estimating that in some cases, the degree of degradation might be greater than 90%.70 In addition, many workers have reported that the sensitivity of detecting a single aberrantly methylated marker might be quite low. Possible explanations for this observation include the fact that a particular tumor suppressor gene may be aberrantly methylated in only a proportion of tumors, and the fact that bisulfite-related DNA degradation might reduce the target DNA available for analysis. Thus, many researchers have used a combination of methylated markers for cancer detection purposes.78 In an attempt to bypass the use of the bisulfite conversion step, Chan and associates investigated the use of methylation-sensitive restriction enzyme digestion followed by real-time PCR detection of the RASSF1 (RASSF1A) gene, which is hypermethylated in a significant proportion of HCC.17 The methylation-sensitive restriction enzyme will not cleave the hypermethylated, tumor-derived RASSF1 sequences while digesting the background nonmethylated sequences of nontumoral origin. This assay shows that hypermethylated RASSF1 sequences are detectable in 93% of the plasma of patients with HCC, in only 58% of chronic carriers of the hepatitis B virus (HBV), and in 8% of healthy controls. Furthermore, for the former two groups, the median plasma concentration of hypermethylated RASSF1A sequences in the cancer group is some 6.5 times higher than that in the HBV carrier group. A possible disadvantage of this approach is the possibility that incomplete digestion by the restriction enzyme will lead to false-positive results. In this regard, quality control steps for assessing the enzyme digestion step are essential. Future studies are needed to investigate the robustness of this approach in a large-scale clinical setting. Another exciting development in plasma-based methylation analysis is the use of BEAMing for methylation analysis, so-called methyl-BEAMing.108 This technology allows the digital analysis of individual bisulfite-converted DNA molecules through emulsion PCR, bead capture, and magnetic separation. This technology allows the detection of one methylated DNA molecule in a background of approximately 5000 unmethylated molecules. Clinical utility of this approach has been demonstrated through detection of the hypermethylated vimentin gene in the plasma of colorectal cancer patients. Several tumor types are associated with viral infection. Examples include the association of HBV infection with HCC, human papillomavirus (HPV) with cervical carcinoma, and EBV infection with NPC. Numerous workers have looked for tumor-associated viral sequences in the plasma or serum of cancer patients. The most convincing data have been reported for detection of EBV DNA in the plasma or serum of NPC patients.119,144 With the use of real-time PCR technology, EBV DNA has been detected in NPC patients with a sensitivity of 96% and a specificity of 93%.119 The plasma concentrations appear to be positively correlated with tumor load and stage, and can serve as a prognosticator.15,104,113,118 Furthermore, measurement of circulating EBV DNA concentrations can allow one to monitor treatment response and to predict tumor relapse.14,115 Preliminary data suggest that detection of circulating EBV DNA is a marker for the screening of NPC.103 It has been shown that circulating EBV DNA in NPC patients consists of short DNA fragments, instead of complete virions.20 EBV has also been associated with certain lymphomas and a small proportion of gastric carcinomas; plasma/serum EBV DNA has been detected in the plasma of a proportion of such patients.99,100,120 A similar approach has been applied to the detection of HPV DNA in the plasma/serum of patients with cervical cancer.* However, the sensitivity of this approach varies greatly from group to group, with several groups reporting very low values. For example, Dong and colleagues reported that only 6.9% of plasma samples obtained from patients with invasive cervical cancer were HPV DNA positive.47 Similarly, Sathish and coworkers reported a plasma positivity rate of only 11.8% in cervical cancer patients.169 Thus, the clinical role, if any, of detecting HPV DNA in the plasma of cervical cancer patients remains unclear at the present time. With the success of detecting tumor-derived DNA molecules in the plasma of cancer patients, some workers have started to explore the detection of tumor-derived RNA in plasma. This was first achieved in 1999 by two groups: through the detection of tyrosinase mRNA in the serum of melanoma patients, and through EBV RNA in the plasma of NPC patients.93,114 These observations were initially rather intriguing, because RNA has been generally regarded as a highly unstable class of molecules. Subsequent work has demonstrated that plasma RNA is particle associated—a configuration that might protect the RNA molecules from degradation in plasma.64,74,149 Indeed, endogenous plasma RNA has been demonstrated to be stable enough to be processed with several hours’ delay from sample collection to preparation. Consistent with its particle-associated nature, plasma RNA has been shown to be degraded rapidly if subjected to detergent treatment (which would disrupt the particles),50 filtration with submicron-size filters,50,149 and high-speed centrifugation.149 Although plasma RNA is stable enough to be detectable by sensitive molecular techniques such as PCR, such molecules nonetheless are fragmented and partially degraded, instead of existing as intact mRNA molecules.13 Indeed, many species of tumor-derived mRNA have now been successfully detected in the plasma and serum of cancer patients. Examples include vascular endothelial growth factor (VEGF) and thymidylate synthase mRNA in the plasma of patients with colorectal carcinoma,65,66 Bmi-1 mRNA and RNA subunits of the telomerase complex in the plasma of breast cancer patients,24,178 and lamin B1 mRNA in the plasma of HCC patients.184 Analogous to plasma DNA, investigators have studied the integrity of plasma RNA in cancer patients. However, the observation is reversed, namely, that cancer patients appear to have lower plasma RNA integrity than do controls.207 A possible explanation of this observation is that the concentration of RNase in the plasma/serum of cancer patients is increased.163,176 Much interest has been focused on micro RNAs (miRNAs), a class of short (19 to 25 nucleotides), single-stranded, non–protein-coding RNA that plays an important role in the regulation of gene expression.86 Chim and associates first reported that cell-free miRNA is detectable in human plasma, using placental miRNA in the plasma of pregnant women as a model system.26 Following this publication, a flurry of reports were published confirming the presence of miRNA in plasma and serum.* These reports further show that plasma/serum miRNA can be used as a biomarker for various types of cancer, including lymphoma,98 prostate cancer,141 ovarian cancer,164 lung cancer,23 and colorectal cancer.147 Chim and colleagues showed that miRNA appeared to be very stable in plasma, indeed even more stable than mRNA in plasma.26 Furthermore, it was shown by these workers that most of the detected plasma miRNA passed through a 0.22 µm filter. This finding suggests that plasma miRNAs are predominantly non–particle associated, or that they are associated with particles smaller than 0.22 µm. This issue requires continued investigation because other workers have reported the presence of miRNA in plasma in microparticles referred to as exosomes or microvesicles.81,160 Demonstration of the presence of tumor-derived DNA in the plasma of cancer patients prompted Lo and coworkers in 1997 to explore whether an analogous phenomenon might also be present in pregnancy (i.e., whether the fetus would release its DNA into the plasma of its mother).122 This analogy is particularly intriguing because there are similarities between the placenta of the fetus and a tumor, with the former being referred to as a pseudomalignant tissue.181 Using a sequence on the Y chromosome as a marker for a male fetus, Lo and associates demonstrated that this DNA was detectable in the plasma and serum of a woman bearing a male fetus, thus demonstrating the existence of fetal DNA in maternal plasma.122 This group of investigators went on to develop a real-time PCR-based assay for measuring the fractional and absolute concentrations of fetal DNA in maternal plasma.126 It has since been reported that fetal DNA represents a mean of 3 to 6% of the DNA that is present in maternal plasma, as measured by PCR-based techniques. With the use of more precise digital PCR-based techniques, the median fractional concentration of fetal DNA in maternal plasma has been found to be some two times higher, with a median value around 10% in the first and second trimesters, and 20% during the third trimester.131 The absolute concentration of circulating fetal DNA increases with gestational age, probably as a consequence of the increase in fetal tissue mass. Following delivery, fetal DNA is cleared very rapidly, with a half-life on the order of 16 minutes.129 The tissue of origin of fetal DNA in maternal plasma has been generally agreed to be the placenta. This conclusion is supported by various findings. First, fetal DNA has been found in maternal plasma even in anembryonic pregnancies, in which no embryo was found, but in which the placenta was present.2 Second, it has been shown that fetal DNA in maternal serum carries the placental karyotype in cases in which the fetus proper and the placenta contain different cytogenetic signatures.60 Finally, fetal DNA in maternal plasma has been shown to carry the DNA methylation signature of the placenta.16,27,28,151 Through the use of a series of quantitative PCR assays with different amplicon lengths, Chan and colleagues showed that circulating fetal DNA in maternal plasma consisted of short DNA fragments.21 These observations are consistent with the suggestion that fetal DNA is released from the placenta into the maternal plasma as a result of apoptosis or necrosis.188 Furthermore, it was shown that circulating fetal DNA was generally shorter in length than the maternally derived DNA in maternal plasma.21 This observation has opened up the possibility of preferentially enriching for fetal DNA by targeting short DNA fragments. One way of doing this is by electrophoresis of plasma DNA fragments.109,112 Another method consists of digital PCR analysis of plasma DNA from maternal plasma using a nested set of PCR primers.132 The nested set of primers would produce amplicons of different sizes. A digital PCR well containing a long (e.g., maternal) template DNA molecule would be predicted to produce both a long and a short amplicon. Conversely, a digital PCR well containing a short (e.g., fetal) DNA molecule would produce only the short amplicon. This has been referred to as digital size selection.132 One advantage of this system is that the size cutoff can be readily changed by designing PCR primers with different amplicon sizes. Rapid clearance of circulating fetal DNA following delivery has prompted some investigators to study the mechanisms of clearance. Botezatu and associates postulated that a proportion of fetal DNA in maternal plasma might be cleared via the urinary system, with some fetal DNA still detectable in maternal urine.10 However, several other groups have been unable to detect fetal DNA in maternal urine, or they did so only at a relatively low proportion, even during the third trimester, or in pathologies associated with increased concentrations of fetal DNA in maternal plasma (e.g., preeclampsia).83,111,135 One possible explanation for the difficulties experienced by these latter groups is that fetal DNA in maternal urine, if present, would be even more fragmented than its counterpart in maternal plasma. Thus, very short amplicons or special methods for urine DNA extraction might be required for such analysis.177 The first diagnostic applications of cell-free fetal DNA in maternal plasma have centered on those that could be achieved through the detection of fetal-derived paternally inherited sequences that are not present in the genome of the pregnant woman (Figure 45-1). One example of such an application is fetal sex determination by the detection of Y chromosomal sequences from a male fetus in maternal plasma. This strategy is useful for the noninvasive prenatal diagnosis of sex-linked disorders.38,82 This approach is also useful for the prenatal investigation of congenital adrenal hyperplasia, because if the fetus can be shown to be male, then prenatal steroid treatment to avoid virilization could be avoided.82,165 Another example of such an application is the prenatal determination of fetal RhD status in pregnancies involving an RhD-negative pregnant woman.52,59,123 The robustness of the fetal sex and RhD applications is high, and these applications have been utilized for clinical purposes in a number of centers in Europe and the United States. Several groups have reported the detection of paternally inherited fetal gene mutations in maternal plasma. Examples include mutations causing achondroplasia,110,168 myotonic dystrophy,3 α-thalassemia,34 β-thalassemia,76 and cystic fibrosis.11,67 For autosomal dominant disorders, such as achondroplasia, the detection of a fetal-derived mutation from the plasma of a woman who does not carry the mutation is indicative of an at-risk fetus (see Figure 45-1). However, for autosomal recessive disorders, early work in this area was focused on families in which the father and mother carried different mutations (see Figure 45-1). Thus, detection of the mutation that the father has passed on to the fetus would only increase the fetal risk of being a compound heterozygous victim of the disease from 25 to 50%. Conversely, if the paternal mutation is shown to be undetectable in maternal plasma, an affected fetus can be excluded (see Figure 45-1).34 Diagnostically, the latter scenario is more useful for patient management than the former scenario. An alternative way is to examine genetic polymorphisms linked to the wild-type (i.e., nonmutant) gene in the father and demonstrate that the fetus has inherited the wild-type paternal marker combination.31,46 A technical challenge for these applications is the need to detect a minority sequence (i.e., the fetal target) that might differ from the majority background (i.e., the maternal background) by as little as a single base pair (e.g., for the detection of a single point mutation, or an SNP allele). Examples of methods that have been developed for this purpose include single-allele base extension46 or allele-specific base extension,191 followed by mass spectrometry or peptide nucleic acid clamping.109 Investigators have combined (1) the previously mentioned size selection strategy for enriching shorter fetal DNA molecules, and (2) such assays for detecting fetal-derived gene mutations or polymorphisms in maternal plasma.109 Another very promising approach for the sensitive and specific detection of fetal-derived gene mutations and polymorphisms is through the use of digital PCR.125,131,132 Thus, by the use of highly diluted DNA samples and multiple digital PCRs, in which each reaction contains a mean of some 0.5 target molecule, most reactions would contain just a single target template molecule or no target molecule. In other words, fetal and maternal DNA molecules derived from the same locus would be distributed into separate PCR wells, and thus would not interfere with each other during the amplification process. This is analogous to the detection of tumor-derived gene mutations using the digital PCR-based approach described earlier in this chapter. The advent of refinements to the digital PCR process, including microfluidics131 and BEAMing,44,45,48 potentially makes this approach more suited to routine clinical application. Another challenge in this field is to extend the application of plasma DNA-based fetal diagnostics beyond the detection of paternally inherited gene mutations or polymorphic alleles that are absent in the maternal genome. For autosomal recessive disease, such a development would allow a complete diagnosis to be made, instead of just limiting the approach to the exclusion of an affected compound heterozygous fetus.31,34 This goal has been achieved through the development of the concept of relative mutation detection (RMD) (see Figure 45-1).132 Thus, for an autosomal recessive disease in which the mother and the father are carriers of the same mutation, the ratio of mutant and wild-type alleles in the parental genomes would be 1 : 1. If the fetus has inherited a mutant allele from both of its parents, then the ratio of the mutant to the wild-type allele in its genome would be 2 : 0. When this fetus then releases its DNA into the maternal plasma, a slight excess of mutant over wild-type alleles in the mixture of genomes is seen in the maternal circulation. This allelic imbalance will be small and correlated with the fractional fetal DNA concentration in maternal plasma. With the use of the superior quantitative precision of digital PCR techniques, such an allele imbalance has been detected successfully in maternal plasma.132 Conversely, if the fetus has inherited one mutant allele from one of its parents and a wild-type allele from the other parent, then the ratio of mutant to wild-type alleles in its genome would also be 1 : 1. Thus, when this fetus then releases its DNA into the maternal plasma, no allelic imbalance would be observed. With future developments in digital PCR technologies and other digital counting techniques (e.g., massively parallel DNA sequencing described later), it is hoped that this approach will enter clinical practice.
Plasma Nucleic Acids
Discovery and Early Work
Circulating DNA as a Tumor Marker
Features
Examples
Total plasma DNA concentration
Elevated in cancer patients over controls
Plasma DNA size
Plasma DNA molecules are longer (higher integrity) in cancer patients than in controls
Loss of heterozygosity
Microsatellite and single-nucleotide polymorphism analysis
Gene mutations
KRAS, APC, TP53, EGFR
Aberrant DNA methylation
P16, SEPT9 (septin 9), APC, GSTP1, RASSF1, rarB2
Tumor-associated viral DNA
HBV, HPV, EBV
Plasma RNA
Tyrosinase, VEGF, thymidylate synthase, BMI1, laminin B1
Plasma RNA integrity
Plasma RNA molecules are more fragmented (reduced integrity) in cancer patients than in controls
Plasma microRNA
Aberrant concentrations of certain microRNA species in a number of malignancies
Measurement of Total Plasma DNA Concentrations
Plasma DNA Size as a Tumor Marker
Detection of Loss of Heterozygosity in Plasma
Detection of Somatic Gene Mutations in Plasma
Detection of Aberrant DNA Methylation in Plasma
Viral DNA in Plasma
Circulating RNA as a Tumor Marker
Fetal Nucleic Acids in Maternal Plasma
Discovery and Basic Biology of Fetal DNA in Maternal Plasma
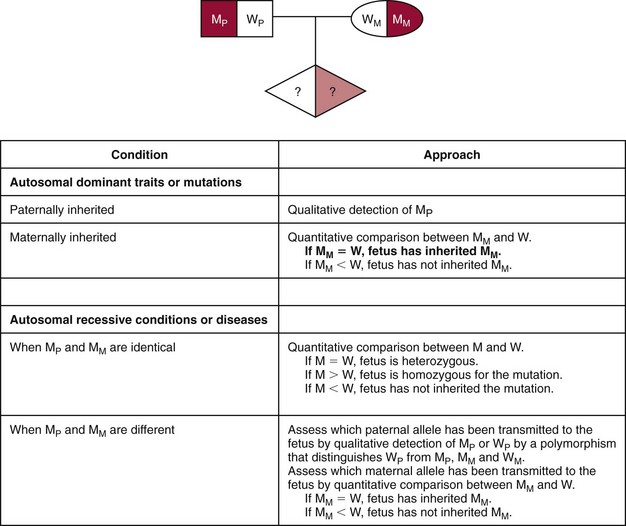
Applications to Sex-Linked Diseases, RhD Genotyping, and Monogenic Disease
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Basicmedical Key
Fastest Basicmedical Insight Engine