
The antiseizure activity of phenytoin is related to its ability to inhibit the repetitive firing of action potentials caused by prolonged depolarization of neurons.7,8 Additionally, phenytoin stops the spread of abnormal discharges from epileptic foci thereby decreasing the spread of seizure activity throughout the brain. Posttetanic potentiation at synaptic junctions are blocked which alters synaptic transmission. At the cellular level, the mechanism of action for phenytoin appears related to its ability to prolong the inactivation of voltage-activated sodium ion channels and reduction of the ability of neurons to fire at high frequencies.
THERAPEUTIC AND TOXIC CONCENTRATIONS
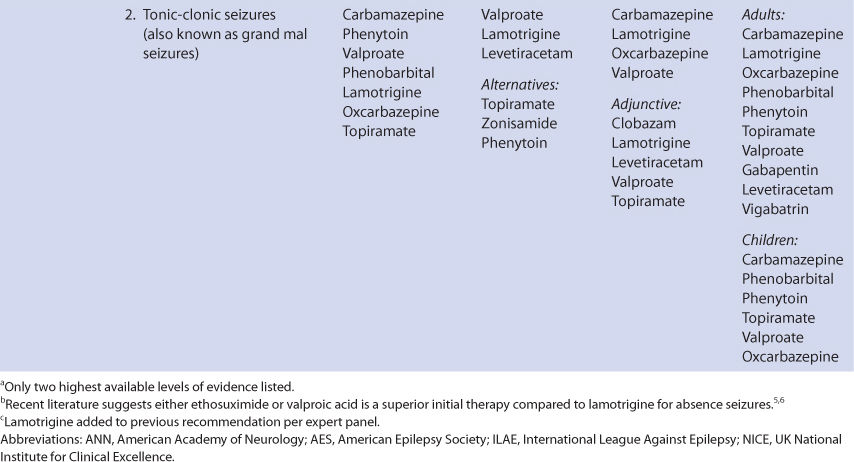
The usual therapeutic range for total (unbound + bound) phenytoin serum concentrations when the drug is used in the treatment of seizures is 10-20 μg/mL. Since phenytoin is highly bound (~90%) to albumin, it is prone to plasma protein–binding displacement due to a large variety of factors. Because of this, unbound or “free” phenytoin concentrations are widely available. Although there is clinical data to support the therapeutic range for total phenytoin concentrations, the suggested therapeutic range for unbound phenytoin concentrations is based on the usual unbound fraction (10%) of phenytoin in individuals with normal plasma protein binding. Thus, the generally accepted therapeutic range for unbound phenytoin concentrations is 1-2 μg/mL, which is simply 10% of the lower and upper bounds for the total concentration range, respectively.
In the upper end of the therapeutic range (>15 μg/mL) some patients will experience minor central nervous system depression side effects such as drowsiness or fatigue.7,8 At total phenytoin concentrations above 20 μg/mL, nystagmus may occur and can be especially prominent upon lateral gaze. When total concentrations exceed 30 μg/mL, ataxia, slurred speech, and/or incoordination similar to ethanol intoxication can be observed. If total phenytoin concentrations are above 40 μg/mL, mental status changes, including decreased mentation, severe confusion or lethargy, and coma are possible. Drug-induced seizure activity has been observed at concentrations over 50-60 μg/mL. Because phenytoin follows nonlinear or saturable metabolism pharmacokinetics, it is possible to attain excessive drug concentrations much easier than for other compounds that follow linear pharmacokinetics. Clinicians should understand that all patients with “toxic” phenytoin serum concentrations in the listed ranges will not exhibit signs or symptoms of phenytoin toxicity. Rather, phenytoin concentrations in the ranges given increase the likelihood that an adverse drug effect will occur.
CLINICAL USEFULNESS OF UNBOUND PHENYTOIN CONCENTRATIONS
Unbound phenytoin concentrations are an extremely useful monitoring tool when used correctly. The relationship between total concentration (C), unbound or “free” concentration (Cf), and unbound or “free” fraction (fB) is: Cf = fBC. For routine therapeutic drug monitoring purposes, total phenytoin serum concentrations are still the mainstream way to gauge therapy with the anticonvulsant. In most patients without known or identifiable plasma protein–binding abnormalities, the unbound fraction of phenytoin will be normal (~10%) and unbound drug concentration measurement is unnecessary. At present, unbound drug concentrations are 50%-100% more expensive than total concentrations, take longer to conduct by the laboratory and have results returned to clinicians, and are not available at all laboratories. Generally, unbound phenytoin serum concentration monitoring should be restricted to those patients with known reasons to have altered drug plasma protein binding. Exceptions to this approach are patients with an augmented or excessive pharmacologic response compared to their total phenytoin concentration. For example, if a patient has a satisfactory anticonvulsant response to a low total phenytoin concentration, one possible reason would be abnormal plasma protein binding (20%) for some unidentified reason, so that even though the total concentration was low (5 μg/mL), a therapeutic unbound concentration was present in the patient (Cf = fBC = 0.2 • 5 μg/mL = 1 μg/mL). Conversely, if a patient has a possible phenytoin-related adverse drug reaction and the total phenytoin concentration is within the therapeutic range, a possible reason could be abnormal protein binding (20%) for an unidentified reason, so that even though the total concentration appeared to be appropriate (15 μg/mL), a toxic unbound concentration was present in the patient (Cf = fBC = 0.2 • 15 μg/mL = 3 μg/mL).
Unbound phenytoin serum concentrations should be measured in patients with factors known to alter phenytoin plasma protein binding. These factors fall into three broad categories: (1) lack of binding protein where there are insufficient plasma concentrations of albumin, (2) displacement of phenytoin from albumin-binding sites by endogenous compounds, and (3) displacement of phenytoin from albumin-binding sites by exogenous compounds (Table 10-2).9–27 When multiple factors that decrease phenytoin plasma protein binding are present in a patient, the free fraction can be as high as 30%-40%.28

Low albumin concentrations, known as hypoalbuminemia, can be found in patients with liver disease or the nephrotic syndrome, pregnant women, cystic fibrosis patients, burn patients, trauma patients, malnourished individuals, and the elderly. Albumin concentrations below 3 g/dL are associated with high phenytoin-unbound fractions in the plasma. Patients with albumin concentrations between 2.5 and 3 g/dL typically have phenytoin-unbound fractions of 15%-20%, while patients with albumin concentrations between 2.0 and 2.5 g/dL often have unbound phenytoin fractions greater than 20%. Albumin is manufactured by the liver so patients with hepatic disease may have difficulty synthesizing the protein. Patients with nephrotic syndrome waste albumin by eliminating it in the urine. Malnourished patients can be so nutritionally deprived that albumin production is impeded. Malnourishment is the reason for hypoalbuminemia in some elderly patients, although there is a general downtrend in albumin concentrations in older patients. While recovering from their injuries, burn and trauma patients can become hypermetabolic and albumin concentrations decrease if enough calories are not supplied during this phase of their disease state. Albumin concentrations may decline during pregnancy as maternal reserves are shifted to the developing fetus and are especially prevalent during the third trimester.
Displacement of phenytoin from plasma protein–binding sites by endogenous substances can occur in patients with hepatic or renal dysfunction. The mechanism is competition for albumin plasma protein–binding sites between the exogenous substances and phenytoin. Bilirubin (a byproduct of heme metabolism) is broken down by the liver, so patients with hepatic disease can have excessive bilirubin concentrations. Total bilirubin concentrations in excess of 2 mg/dL are associated with abnormal phenytoin plasma protein binding. End-stage renal disease patients (creatinine clearance <10-15 mL/min) with uremia (blood urea nitrogen concentrations >80-100 mg/dL) accumulate unidentified compound(s) in their blood that displace phenytoin from plasma protein–binding sites. Abnormal phenytoin binding persists in these patients even when dialysis procedures are instituted.
Phenytoin plasma protein–binding displacement can also occur due to exogenously administered compounds such as drugs. In this case, the mechanism is competition for albumin-binding sites between phenytoin and other agents. Other drugs that are highly bound to albumin and cause plasma protein–binding displacement drug interactions with phenytoin include warfarin, valproic acid, aspirin (>2 g/d), and some highly bound nonsteroidal anti-inflammatory agents.
Once the free fraction (fB) has been determined for a patient with altered phenytoin plasma protein binding (fB = Cf/C, where C is the total concentration and Cf is the unbound concentration), it is often not necessary to obtain additional unbound drug concentrations. If the situations that caused altered plasma protein binding are stable (albumin or bilirubin concentration, hepatic or renal function, other drug doses, etc), total phenytoin concentrations can be converted to concurrent unbound values and used for therapeutic drug-monitoring purposes. For example, an end-stage renal failure patient is receiving phenytoin therapy as well as valproic acid and warfarin. The concurrently measured total and unbound phenytoin concentrations are 5 μg/mL and 1.5 μg/mL, respectively, yielding an unbound fraction of 30% (fB = Cf/C = 1.5 μg/mL / 5 μg/mL = 0.30). The next day, a total phenytoin concentration is measured and equals 6 μg/mL. The estimated unbound concentration using this information would be 1.8 μg/mL: Cf = fBC = 0.30 • 6 μg/mL = 1.8 μg/mL. Of course, if the disease state status or drug therapy changes, a new unbound phenytoin fraction will be present and need to be remeasured using an unbound/total phenytoin concentration pair.
When unbound phenytoin concentrations are unavailable, several methods have been suggested to estimate the value or a surrogate measure of the value. The most common surrogate is an estimation of the equivalent total phenytoin concentration that would provide the same unbound phenytoin concentration if the patient had a normal unbound fraction value of 10%. These calculations “normalize” the total phenytoin concentration so that it can be compared to the usual phenytoin therapeutic range of 10-20 μg/mL and used for dosage adjustment purposes. The equation for hypoalbuminemia is: CNormalBinding = C/(X • Alb + 0.1), where CNormalBinding is the normalized total phenytoin concentration in μg/mL, C is the actual measured phenytoin concentration in μg/mL, X is a constant equal to 0.2 if protein-binding measurements were conducted at 37°C or 0.25 if conducted at 25°C, and Alb is the albumin concentration in g/dL.29,30 If the patient has end-stage renal disease (creatinine clearance <10-15 mL/min), the same equation is used with a different constant value (X = 0.1).29 (Note: in most experimental laboratories, protein binding is determined at normal body temperature [37°C], in most clinical laboratories, protein binding is determined at room temperature [25°C]). Because these methods assume that the normal unbound fraction of phenytoin is 10%, the estimated unbound phenytoin concentration (CfEST) is computed using the following formula: (CfEST) = 0.1 CNormalBinding. A different approach is taken by the equations used for patients with concurrent valproic acid administration. In this case, the unbound phenytoin concentration (CfEST) is estimated using simultaneously measured total phenytoin (PHT in μg/mL) and valproic acid (VPA in μg/mL) concentrations: CfEST = (0.095 + 0.001 • VPA)PHT.31,32 This value is compared to the usual therapeutic range for unbound phenytoin concentrations (1-2 μg/mL) and used for dosage adjustment purposes. It should be noted that these equations only provide estimates of their respective concentrations, and actual unbound phenytoin concentrations should be measured whenever possible in patients with suspected abnormal phenytoin plasma protein binding.
EXAMPLE 1 
JM is an epileptic patient being treated with phenytoin. He has hypoalbuminemia (albumin = 2.2 g/dL) and normal renal function (creatinine clearance = 90 mL/min). His total phenytoin concentration is 7.5 μg/mL. Assuming that any unbound concentrations performed by the clinical laboratory will be conducted at 25°C, compute an estimated normalized phenytoin concentration for this patient.
1. Choose the appropriate equation to estimate normalized total phenytoin concentration at the appropriate temperature.

This patient’s estimated normalized total phenytoin concentration is expected to provide an unbound concentration equivalent to a total phenytoin concentration of 11.5 μg/mL for a patient with normal drug protein binding (CfEST = 1.2 μg/mL). Because the estimated total value is within the therapeutic range of 10-20 μg/mL, it is likely that the patient has an unbound phenytoin concentration within the therapeutic range. If possible, this should be confirmed by obtaining an actual, measured unbound phenytoin concentration.
EXAMPLE 2 
LM is an epileptic patient being treated with phenytoin. He has hypoalbuminemia (albumin = 2.2 g/dL) and poor renal function (creatinine clearance = 10 mL/min). His total phenytoin concentration is 7.5 μg/mL. Compute an estimated normalized phenytoin concentration for this patient.
1. Choose appropriate equation to estimate normalized total phenytoin concentration.

This patient’s estimated normalized total phenytoin concentration is expected to provide an unbound concentration equivalent to a total phenytoin concentration of 23.4 μg/mL for a patient with normal drug protein binding (CfEST = 2.3 μg/mL). Because the estimated total value is above the therapeutic range of 10-20 μg/mL, it is likely that the patient has an unbound phenytoin concentration above the therapeutic range. If possible, this should be confirmed by obtaining an actual, measured unbound phenytoin concentration.

PM is an epileptic patient being treated with phenytoin and valproic acid. He has a normal albumin concentration (albumin = 4.2 g/dL) and normal renal function (creatinine clearance = 90 mL/min). His steady-state total phenytoin and valproic acid concentrations are 7.5 μg/mL and 100 μg/mL, respectively. Compute an estimated unbound phenytoin concentration for this patient.
1. Choose appropriate equation to estimate unbound phenytoin concentration.

This patient’s estimated unbound phenytoin concentration is expected to be within the therapeutic range for unbound concentrations. If possible, this should be confirmed by obtaining an actual, measured unbound phenytoin concentration.
CLINICAL MONITORING PARAMETERS
The goal of therapy with anticonvulsants is to reduce seizure frequency and maximize quality of life with a minimum of adverse drug effects.7 While it is desirable to entirely abolish all seizure episodes, it may not be possible to accomplish this in many patients. Patients should be monitored for concentration-related side effects (drowsiness, fatigue, nystagmus, ataxia, slurred speech, incoordination, mental status changes, decreased mentation, confusion, lethargy, coma) as well as adverse reactions associated with long-term use (behavioral changes, cerebellar syndrome, connective tissue changes, coarse facies, skin thickening, folate deficiency, gingival hyperplasia, lymphadenopathy, hirsutism, osteomalacia). Idiosyncratic side effects include skin rash, Stevens-Johnson syndrome, bone marrow suppression, systemic lupus-like reactions, and hepatitis.
Phenytoin serum concentrations should be measured in most patients. Because epilepsy is an episodic disease state, patients do not experience seizures on a continuous basis. Thus, during dosage titration, it is difficult to tell if the patient is responding to drug therapy or simply is not experiencing any abnormal central nervous system discharges at that time. Phenytoin serum concentrations are also valuable tools to avoid adverse drug effects. Patients are more likely to accept drug therapy if adverse reactions are held to the absolute minimum. Because phenytoin follows nonlinear or saturable pharmacokinetics, it is fairly easy to attain toxic concentrations with modest changes in drug dose.
BASIC CLINICAL PHARMACOKINETIC PARAMETERS
Phenytoin is primarily eliminated by hepatic metabolism (>95%). Hepatic metabolism is mainly via the CYP2C9 enzyme system with a smaller amount metabolized by CYP2C19. About 5% of a phenytoin dose is recovered in the urine as unchanged drug. Phenytoin follows Michaelis-Menten or saturable pharmacokinetics.33–35 This is the type of nonlinear pharmacokinetics that occurs when the number of drug molecules overwhelms or saturates the enzyme’s ability to metabolize the drug. When this occurs, steady-state drug serum concentrations increase in a disproportionate manner after a dosage increase (Figure 10-1). In this case, the rate of drug removal is described by the classic Michaelis-Menten relationship that is used for all enzyme systems: rate of metabolism = (Vmax • C)/(Km + C), where Vmax is the maximum rate of metabolism in mg/d, C is the phenytoin concentration in mg/L or μg/mL, and Km is the substrate concentration in mg/L or μg/mL where the rate of metabolism = Vmax/2.
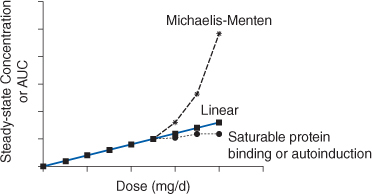
FIGURE 10-1 If a drug follows linear pharmacokinetics, Css or AUC increases proportionally with dose resulting in a straight line on the plot. Nonlinear pharmacokinetics occurs when the Css or AUC versus dose plot results in something other than a straight line. If a drug follows Michaelis-Menten pharmacokinetics (eg, phenytoin, aspirin), as steady-state drug concentrations approach Km serum concentrations increase more than expected due to dose increases. If a drug follows nonlinear protein binding (eg, valproic acid, disopyramide) or demonstrates autoinduction (eg, carbamazepine), total steady-state drug concentrations increase less than expected as dose increases.
The clinical implication of Michaelis-Menten pharmacokinetics is that the clearance of phenytoin is not a constant as it is with linear pharmacokinetics, but is concentration or dose dependent. As the dose or concentration of phenytoin increases, the clearance rate (Cl) decreases as the enzyme approaches saturable conditions: Cl = Vmax/(Km + C). This is the reason concentrations increase disproportionately after a phenytoin dosage increase. For example, phenytoin follows saturable pharmacokinetics with average Michaelis-Menten constants of Vmax = 500 mg/d and Km = 4 mg/L. The therapeutic range of phenytoin is 10-20 μg/mL. As the steady-state concentration of phenytoin increases from 10 μg/mL to 20 μg/mL, clearance decreases from 36 L/d to 21 L/d: Cl = Vmax/(Km + C); Cl = (500 mg/d)/(4 mg/L + 10 mg/L) = 36 L/d; Cl = (500 mg/d)/(4 mg/L + 20 mg/L) = 21 L/d (note: μg/mL = mg/L and this substitution was directly made to avoid unnecessary unit conversion). Unfortunately, there is so much interpatient variability in Michaelis-Menten pharmacokinetic parameters for phenytoin (typically Vmax = 100-1000 mg/d and Km = 1-15 μg/mL) that dosing the drug is extremely difficult.
Phenytoin volume of distribution (V = 0.7 L/kg) is unaffected by saturable metabolism and is still determined by the physiological volume of blood (VB) and tissues (VT) as well as the unbound concentration of drug in the blood (fB) and tissues (fT): V = VB+ (fB/fT)VT. Also, half-life (t1/2) is still related to clearance and volume of distribution using the same equation as for linear pharmacokinetics: t1/2 = (0.693 • V)/Cl. However, since clearance is dose or concentration dependent, half-life also changes with phenytoin dosage or concentration changes. As doses or concentrations increase for a drug that follows Michaelis-Menten pharmacokinetics, clearance decreases and half-life becomes longer for the drug: ↑t1/2 = (0.693 • V)/↓Cl. Using the above example for clearance and the volume of distribution for a 70-kg person (V = 0.7 L/kg • 70 kg ≈ 50 L), half-life changes from 1 day [t1/2 = (0.693 • V)/Cl = (0.693 • 50 L)/36 L/d = 1 d] to 1.7 days [t1/2 = (0.693 • 50 L)/ 21 L/d = 1.7 d] as phenytoin serum concentrations increase from 10 μg/mL to 20 μg/mL. The clinical implication of this finding is that the time to steady-state (3-5 t1/2) is longer as the dose or concentration is increased for phenytoin. On average, the time to steady-state serum concentrations is approximately 5 days at a dosage rate of 300 mg/d and 15 days at a dosage rate of 400 mg/d.34
Under steady-state conditions, the rate of drug administration equals the rate of drug removal.36 Therefore, the Michaelis-Menten equation can be used to compute the maintenance dose (MD in mg/d) required to achieve a target steady-state phenytoin serum concentration (Css in μg/mL or mg/L):
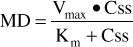
Or, solved for Css:
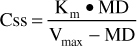
When phenytoin steady-state concentrations are far below the Km value for a patient, this equation simplifies to: MD = (Vmax/Km)Css or, since Vmax/Km is a constant, MD = Cl • Css. Therefore, when Km >> Css, phenytoin follows linear pharmacokinetics. When phenytoin steady-state concentrations are far above the Km value for a patient, the rate of metabolism becomes a constant equal to Vmax. Under these conditions, only a fixed amount of phenytoin is metabolized per day because the enzyme system is completely saturated and cannot increase its metabolic capacity. This situation is also known as zero-order pharmacokinetics. First-order pharmacokinetics is another name for linear pharmacokinetics.
For parenteral use, phenytoin is available in two different dosage forms. Phenytoin sodium, the sodium salt of phenytoin, contains 92% phenytoin by weight. Even though it is a salt of phenytoin, the drug is still relatively insoluble in water. To facilitate dissolution, ethanol and propylene glycol are added to the vehicle, and the pH of the solution is adjusted to between 10 and 12. When given intramuscularly, phenytoin sodium injections are very painful.37 Some of the drug probably precipitates in the muscle injection site and this results in prolonged absorption of drug over several days. When given intravenously, injection rates should not exceed 50 mg/min to avoid hypotension. Even at lower infusion rates, profound hypotension can result in patients with unstable blood pressure or shock. Phenytoin sodium injection can be given by slow intravenous push of undiluted drug, or added to normal saline at a concentration of 10 mg/mL or less and infused <50 mg/min. When added to normal saline, the drug should be given as soon as possible after being mixed to avoid precipitation, and a 0.22-μm inline filter should be used to remove any drug crystals before they reach the patient.
To avoid many of the problems associated with phenytoin sodium injection, a water-soluble phosphate ester prodrug of phenytoin, fosphenytoin, has been developed. Conversion of fosphenytoin to phenytoin is rapid, with a fosphenytoin half-life of approximately 15 minutes.38 To avoid confusion, fosphenytoin is prescribed in terms of phenytoin sodium equivalents (PE). Thus, 100 mg PE of fosphenytoin is equivalent to 100 mg of phenytoin sodium. Hypotension during intravenous administration of fosphenytoin is much less of a problem than with phenytoin sodium. The maximal intravenous infusion rate is 150 mg PE/min. Transient pruritus and paresthesia are associated with this route of administration. Intramuscular absorption is rapid with a peak concentration that occurs about 30 minutes after injection, and bioavailability via this route of administration is 100%. However, fosphenytoin is much more expensive that phenytoin sodium injection, and this has limited its widespread use. Because of this, most clinicians have reserved fosphenytoin use to patients requiring intramuscular phenytoin or to patients with unstable or low blood pressure requiring intravenous phenytoin therapy.
For oral use, capsules contain phenytoin sodium (92% phenytoin, by weight) while tablets and suspension contain phenytoin. Phenytoin sodium capsules are labeled as extended phenytoin sodium capsules or prompt phenytoin capsules. Extended phenytoin capsules release phenytoin slowly from the gastrointestinal tract into the systemic circulation. The extended-release characteristics of this dosage form are due to the slow dissolution of the drug in gastric juices and not the result of extended-release dosage form technology. Prompt phenytoin sodium capsules are absorbed fairly quickly from the gastrointestinal tract because they contain microcrystalline phenytoin sodium, which dissolves quicker in gastric juices. As a result of their sustained-release properties, phenytoin doses given as extended phenytoin sodium capsules can be given every once or twice daily, but prompt phenytoin sodium capsules must be given multiple times daily. Extended phenytoin sodium capsules are available in 30-mg, 100-mg, 200-mg, and 300-mg strengths. Because phenytoin follows nonlinear pharmacokinetics, even subtle differences in bioavailability may alter steady-state serum concentrations.39,40 Any time therapy is changed from one brand of phenytoin to another, clinicians should carefully monitor patients for any changes in seizure frequency, adverse effects, and steady-state concentration.
Phenytoin tablets (50 mg, chewable) and suspension (125 mg/5 mL) for oral use are available as the acid form of the drug. Both the tablet and suspension dosage forms are absorbed more rapidly than extended phenytoin sodium capsules, and once daily dosing with these may not be possible in some patients. The suspension is thick, and the drug is difficult to disperse evenly throughout the liquid. If not shaken well before dispensing a dose, the drug can flocculate out into the bottom of the bottle. When this occurs, phenytoin concentrations near the top of the bottle will be less than average, and doses given when the bottle is ⅔ or more full will contain less phenytoin. Conversely, phenytoin concentrations near the bottom of the bottle will be greater than average, and doses given when the bottle is ⅓ or less full will contain more phenytoin. This problem can be avoided to a large extent if the dispensing pharmacist shakes the bottle very well (several minutes) before giving to the patient.
For most drugs, the 8% difference in dose between dosage forms containing phenytoin (suspension and tablets, 100 mg = 100 mg phenytoin) and phenytoin sodium (capsules and injection, 100 mg = 92 mg phenytoin) would be trivial and could easily be ignored. However, because phenytoin follows nonlinear pharmacokinetics, an 8% difference in dose can result in major changes in phenytoin serum concentrations. For example, if a patient is stabilized on a dose of intravenous phenytoin sodium 300 mg/d (300 mg/d phenytoin sodium × 0.92 = 276 mg phenytoin) with a steady-state concentration of 17 μg/mL, switching the patient to phenytoin suspension 300 mg/d could result in steady-state phenytoin concentrations exceeding 20 μg/mL (15%-30% increase or more) and result in toxicity. Conversely, if a different patient is stabilized on a dose of phenytoin suspension 300 mg/d with a steady-state concentration of 12 μg/mL, switching the patient to intravenous phenytoin sodium 300 mg/d (300 mg/d phenytoin sodium × 0.92 = 276 mg phenytoin) could result in steady-state phenytoin concentrations below 10 μg/mL (15%-30% decrease or more) and result in loss of efficacy. Usually, phenytoin doses are not fine-tuned to the point of directly accounting for the difference in phenytoin content (ie, 276 mg of phenytoin suspension would not be prescribed for the patient receiving 300 mg of phenytoin sodium injection). Rather, clinicians are aware that when phenytoin dosage forms are changed, phenytoin content may change and anticipate that the drug concentration may increase or decrease because of this. Because of this, most individuals recheck phenytoin serum concentrations after a dosage form change is instituted.
The oral bioavailability of phenytoin is very good for capsule, tablet, and suspension dosage forms and approximates 100%.41–44 At larger amounts, there is some dose dependency on absorption characteristics.45 Single oral doses of 800 mg or more produce longer times for maximal concentrations to occur (Tmax) and decreased bioavailability. Since larger oral doses also produce a higher incidence of gastrointestinal side effects (primarily nausea and vomiting due to local irritation), it is prudent to break maintenance doses larger than 800 mg/d into multiple doses. If oral phenytoin loading doses are given, a common total dose is 1000 mg given as 400 mg, 300 mg, and 300 mg separated by 2- to 6-hour time intervals. Enteral feedings given by nasogastric tube interfere with phenytoin absorption.46–49 Possible mechanisms include decreased gastrointestinal transit time which reduces absorption contact time, binding of phenytoin to proteins contained in the feedings, and adherence of phenytoin to the lumen of the feeding tube. The solution to this problem is to stop the feedings, when possible, for 1-2 hours before and after phenytoin administration, and increase the oral phenytoin dose.48 It is not unusual for phenytoin oral dosage requirements to double or triple while the patient receives concurrent nasogastric feedings (eg, usual dose of 300-400 mg/d increasing to 600-1200 mg/d while receiving nasogastric feedings). Of course, intravenous or intramuscular phenytoin or fosphenytoin doses could also be substituted while nasogastric feedings were being administered. Although poorly documented, phenytoin oral malabsorption may also occur in patients with severe diarrhea, malabsorption syndromes, or gastric resection.
The typical recommended loading dose for phenytoin is 15-20 mg/kg resulting in 1000 mg for most adult patients. Usual initial maintenance doses are 5-10 mg/kg/d for children (6 months to 16 years old) and 4-6 mg/kg/d for adults. For adults, the most prescribed dose is 300-400 mg/d of phenytoin. Because of an increased incidence of adverse effects in older patients (>65 years old), many clinicians prescribe a maximum of 200 mg/d as an initial dose for these individuals.50,51
IMPACT OF ALTERED PLASMA PROTEIN BINDING ON PHENYTOIN PHARMACOKINETICS
The pharmacokinetic alterations that occur with altered plasma protein binding result in complex changes for total and unbound steady-state phenytoin concentrations and drug response. As previously discussed (see Chapter 3, Drug Dosing in Special Populations: Renal and Hepatic Disease, Dialysis, Heart Failure, Obesity, and Drug Interactions), hepatic drug metabolism is described by the following equation:
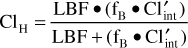
where LBF is liver blood flow, fB is the fraction of unbound drug in the blood, and Cl′int is intrinsic clearance. For drugs such as phenytoin with a low hepatic extraction ratio (≤30%), the numeric value of liver blood flow is much greater than the product of unbound fraction of drug in the blood and the intrinsic clearance of the compound (LBF >> fB • Cl′int), and the sum in the denominator of the hepatic clearance equation is almost equal to liver blood flow [LBF ≈ LBF + (fB • Cl′int)]. When this substitution is made into the hepatic clearance equation, hepatic clearance is equal to the product of free fraction in the blood and the intrinsic clearance of the drug for a drug with a low hepatic extraction ratio:
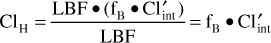
In order to illustrate the differences that may occur in steady-state drug concentrations and pharmacologic effects for patients with altered phenytoin plasma protein binding, a graphical technique will be used (Figure 10-2A). The example assumes that phenytoin is being given to a patient as a continuous intravenous infusion, and that all physiologic, pharmacokinetic, and drug effect parameters (shown on the y-axis) are initially stable. However, the same changes occur for average total and unbound steady-state concentrations when the drug is given on a continuous dosage schedule (every 8, 12, 24, etc hours) or orally. On the x-axis, an arrow indicates that phenytoin plasma protein binding decreases and unbound fraction increases in the patient; an assumption made for this illustration is that any changes in the parameters are instantaneous. An increase in the parameter is denoted as an uptick in the line while a decrease in the parameter is shown as a downtick in the line.
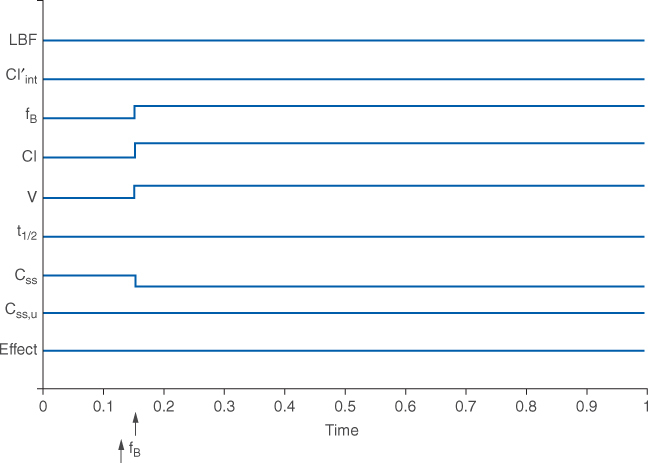
FIGURE 10-2A Schematic representation of physiologic (LBF = liver blood flow, Cl′int = intrinsic or unbound clearance, fB = unbound fraction of drug in blood/plasma), pharmacokinetic (Cl = clearance; V = volume of distribution; t1/2 = half-life; Css = total steady-state drug concentration; Css, u = unbound steady-state drug concentration), and pharmacodynamic (Effect = pharmacodynamic effect) changes that occur with decreased protein binding of phenytoin (arrow denotes ↑fB).
For a drug with a low hepatic extraction ratio, plasma protein–binding displacement drug interactions cause major pharmacokinetic alterations but are not clinically significant because the pharmacologic effect of the drug does not change (Figure 10-2A). Because the clearance of the drug is dependent on the fraction of unbound drug in the blood and intrinsic clearance for a low hepatic extraction ratio agent, a decrease in plasma protein binding and increase in unbound fraction will increase clearance (↑Cl = ↑fBCl′int) and volume of distribution [↑V = VB + (↑fB/fT)VT]. Since half-life depends on clearance and volume of distribution, it is likely that because both increase, half-life will not substantially change [t1/2 = (0.693 • ↑V)/↑Cl]. However, it is possible that if either clearance or volume of distribution changes disproportionately, half-life will change. The total steady-state concentration will decline because of the increase in clearance (↓Css = k0/↑Cl, where k0 is the infusion rate of drug). But, the unbound steady-state concentration will remain unaltered because the free fraction of drug in the blood is higher than it was before the increase in unbound fraction occurred (Css, u = ↑fB↓Css). The pharmacologic effect of the drug does not change because the free concentration of drug in the blood is unchanged. This can be an unexpected outcome for the decrease in plasma protein binding, especially because the total steady-state concentration of the drug decreased. Clinicians need to be on the outlook for situations like this because the total drug concentration (bound + unbound) can be misleading and cause an unwarranted increase in drug dosage. Unbound drug concentrations should be used to convince clinicians that a drug dosage increase is not needed even though total concentrations decline as a result of this interaction.
EFFECTS OF DISEASE STATES AND CONDITIONS ON PHARMACOKINETICS AND DOSING
Measurement of Vmax and Km for phenytoin is very difficult to accomplish for research or clinical purposes. Because of this, the effects of disease states and conditions on these parameters are largely unknown. By necessity, this discussion must be done in qualitative terms for phenytoin. Adults without the disease states and conditions given later in this section, with normal liver and renal function as well as normal plasma protein binding (~90%), have an average phenytoin Vmax of 7 mg/kg/d (range: 1.5-14 mg/kg/d) and Km of 4 μg/mL (range: 1-15 μg/mL).35 Michaelis-Menten parameters for younger children (6 months-6 years) are Vmax = 12 mg/kg/d and Km = 6 μg/mL while for older children (7-16 years) Vmax = 9 mg/kg/d and Km = 6 μg/mL.52–58 The most difficult and frustrating aspect of phenytoin dosage determination is the 10-15-fold variation in Michaelis-Menten pharmacokinetic parameters which creates a huge amount of variability in dose requirements. An individualized dosage regimen for each patient prescribed phenytoin must be determined to accomplish therapeutic goals. Unfortunately, measurement of Vmax and Km for phenytoin is very difficult to accomplish for research or clinical purposes. Because of this, the effects of disease states and conditions on these parameters are largely unknown. By necessity, this discussion must be done in qualitative terms for phenytoin.
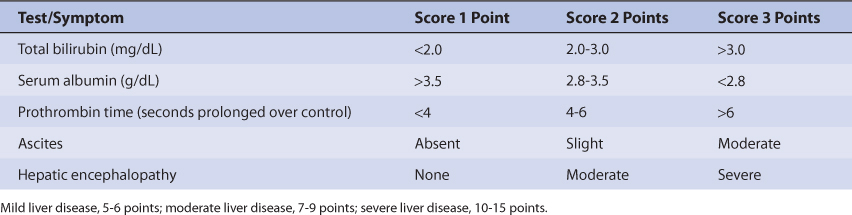
Patients with liver cirrhosis or acute hepatitis have reduced phenytoin clearance because of destruction of liver parenchyma. This loss of functional hepatic cells reduces the amount of CYP2C9 and CYP2C19 available to metabolize the drug and decreases Vmax. The volume of distribution is larger because of reduced plasma protein binding. Protein binding is reduced and unbound fraction is increased due to hypoalbuminemia and/or hyperbilirubinemia (especially albumin ≤3 g/dL and/or total bilirubin ≥2 mg/dL). However, the effects that liver disease have on phenytoin pharmacokinetics are highly variable and difficult to accurately predict. It is possible for a patient with liver disease to have relatively normal or grossly abnormal phenytoin clearance and volume of distribution. For example, a liver disease patient who has relatively normal albumin and bilirubin concentrations can have a normal volume of distribution for phenytoin. An index of liver dysfunction can be gained by applying the Child-Pugh clinical classification system to the patient (Table 10-3).59 Child-Pugh scores are completely discussed in Chapter 3 (Drug Dosing in Special Populations: Renal and Hepatic Disease, Dialysis, Heart Failure, Obesity, and Drug Interactions), but will be briefly discussed here. The Child-Pugh score consists of five laboratory tests or clinical symptoms: serum albumin, total bilirubin, prothrombin time, ascites, and hepatic encephalopathy. Each of these areas is given a score of 1 (normal) to 3 (severely abnormal; see Table 10-3), and the scores for the five areas are summed. The Child-Pugh score for a patient with normal liver function is 5 while the score for a patient with grossly abnormal serum albumin, total bilirubin, and prothrombin time values in addition to severe ascites and hepatic encephalopathy is 15. A Child-Pugh score greater than 8 is grounds for a decrease of 25%-50% in the initial daily drug dose for phenytoin. As in any patient with or without liver dysfunction, initial doses are meant as starting points for dosage titration based on patient response and avoidance of adverse effects. Phenytoin serum concentrations and the presence of adverse drug effects should be monitored frequently in patients with liver cirrhosis.
Other patients are also prone to hypoalbuminemia, including patients with the nephrotic syndrome, cystic fibrosis patients, and malnourished individuals. Unbound phenytoin concentration monitoring should be considered in these patients especially when albumin concentrations are ≤3 g/dL. High bilirubin concentrations can also be found in patients with biliary tract obstruction or hemolysis. Unbound phenytoin concentration monitoring should be considered in these patients especially when total bilirubin concentrations are ≥2 mg/dL.
Trauma and burn patients have an increased ability to metabolize phenytoin beginning 3-7 days after their initial injury.60,61 At this time, these patients become hypermetabolic in order to repair damaged tissue, and the Vmax for phenytoin increases due to this general increase in metabolic rate. If caloric needs are not met during this phase of recovery for trauma patients, many become hypoalbuminemic, and phenytoin plasma protein binding decreases resulting in an increased unbound fraction. Phenytoin dosage requirements are increased while trauma patients are in their hypermetabolic phase, and unbound concentration monitoring is indicated when patients have low albumin concentrations (especially for albumin levels ≤3 g/dL).
Pregnant women taking phenytoin have increased dosage requirements, particularly during the third trimester (>26 weeks).9,10,62–66 There are several reasons for this change, including malabsorption of drug resulting in decreased bioavailability, increased metabolism of phenytoin, and decreased protein binding due to low albumin concentrations. Aggressive drug serum concentration monitoring, including the measurement of unbound phenytoin concentrations if the patient is hypoalbuminemic, is necessary to avoid seizures and subsequent harm to the unborn fetus. An additional concern when administering phenytoin to pregnant patients is the development of fetal hydantoin syndrome by the baby.
Elderly individuals over the age of 65 years have a decreased capacity to metabolize phenytoin, possibly due to age-related losses of liver parenchyma resulting in decreased amounts of CYP2C9 and CYP2C19.50,51 Older patients also may have hypoalbuminemia with resulting decreases in plasma protein binding and increases in unbound fraction.26,27 Many elderly patients also seem to have an increased propensity for central nervous system side effects due to phenytoin, and because of these pharmacokinetic and pharmacodynamic changes, clinicians tend to prescribe lower initial phenytoin doses for older patients (~200 mg/d).
End-stage renal disease patients with creatinine clearances <10-15 mL/min have an unidentified substance in their blood that displaces phenytoin from its plasma protein–binding sites.19–23,25 This unknown compound is not removed by dialysis.24 In addition to this, these patients tend to have hypoalbuminemia which increases the unbound fraction of phenytoin even further. Unbound phenytoin serum concentration monitoring is very helpful in determining dosage requirements for renal failure patients. Other patients are also prone to hypoalbuminemia, including patients with the nephrotic syndrome, cystic fibrosis patients, and malnourished individuals. High bilirubin concentrations can also be found in patients with biliary tract obstruction or hemolysis. Unbound phenytoin concentration monitoring should be considered in these patients especially when albumin concentrations are ≤3 g/dL or total bilirubin concentrations are ≥2 mg/dL.
Hemodialysis does not remove enough phenytoin that supplemental postdialysis doses are necessary.66 The typical sieving coefficient during hemoperfusion for phenytoin is 0.45, so in some cases supplemental phenytoin doses could be needed.68,69 Because of pharmacokinetic variability, check phenytoin concentrations in patients receiving hemoperfusion.
The ratio between simultaneous breast milk and plasma areas under the curve averaged 0.13.70 The mean ratio between breast milk and plasma concentration determined at various times during a dosage interval is 0.28.71
DRUG INTERACTIONS
Because phenytoin is so highly liver metabolized by CYP2C9 and CYP2C19, it is prone to drug interactions that inhibit hepatic microsomal enzymes.72,73 Cimetidine, valproic acid, amiodarone, chloramphenicol, isoniazid, disulfiram, and omeprazole have been reported to inhibit phenytoin metabolism and increase phenytoin serum concentrations. Phenytoin is also a broad-based hepatic enzyme inducer affecting most cytochrome P-450 systems. Drugs with narrow therapeutic ranges that can have their metabolism increased by concurrent phenytoin administration include carbamazepine, phenobarbital, cyclosporin, tacrolimus, and warfarin. When phenytoin therapy is added to the medication regimen for a patient, a comprehensive review for drug interactions should be conducted. Valproic acid, aspirin (>2 g/d), some highly protein-bound nonsteroidal anti-inflammatory drugs, and warfarin can displace phenytoin from plasma protein–binding sites necessitating monitoring of unbound phenytoin concentrations.
The drug interaction between valproic acid and phenytoin deserves special examination because of its complexity and because these two agents are regularly used together for the treatment of seizures.11–14 The drug interaction involves the plasma protein–binding displacement and intrinsic clearance inhibition of phenytoin by valproic acid. What makes this interaction so difficult to detect and understand is that these two changes do not occur simultaneously, so the impression left by the drug interaction depends on when in time it is observed in a patient. For example, a patient is stabilized on phenytoin therapy (Figure 10-2B), but because adequate control of seizures has not been attained, valproic acid is added to the regimen. As valproic acid concentrations accumulate, the first interaction observed is phenytoin plasma protein binding as the two drugs compete for binding sites on albumin. The result of this portion of the drug interaction is an increase in phenytoin unbound fraction and a decrease in phenytoin total serum concentration, but the unbound phenytoin serum concentration remains the same. As valproic acid serum concentrations achieve steady-state conditions, the higher concentrations of the drug bathe the hepatic microsomal enzyme system and inhibit the intrinsic clearance of phenytoin. This portion of the interaction decreases intrinsic clearance and hepatic clearance for phenytoin, so both unbound and total phenytoin concentrations increase. When phenytoin concentrations finally equilibrate and reach steady-state under the new plasma protein binding and intrinsic clearance conditions imposed by concurrent valproic acid therapy, the total concentration of phenytoin is often times at about the same level as before the drug interaction occurred, but unbound phenytoin concentrations are much higher. If only total phenytoin concentrations are measured at this point in time, clinicians will be under the impression that total concentrations did not change and no drug interaction occurred. However, if unbound phenytoin concentrations are simultaneously measured, it will be found that these concentrations have risen and that the phenytoin unbound fraction is twice or more (≥20%) of the baseline amount. In this situation, the patient may have unbound phenytoin concentrations that are toxic and a decrease in phenytoin dosage may be in order.
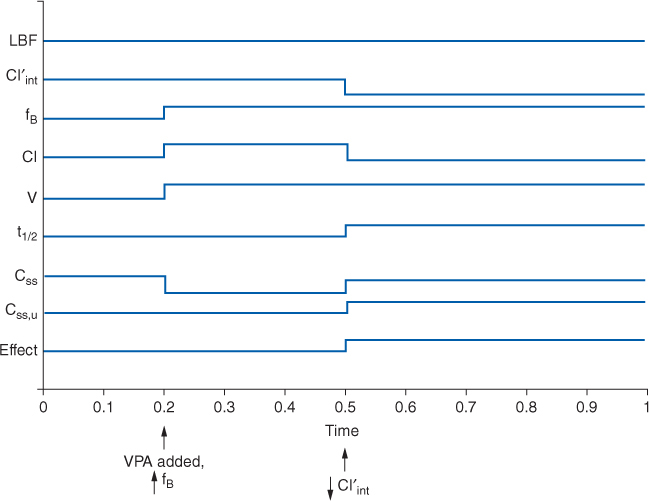
FIGURE 10-2B Schematic representation of the effect of initiating valproic acid (VPA) treatment in an individual stabilized on phenytoin therapy (see Figure 10-2A legend for symbol definition). Initially, valproic acid decreases phenytoin plasma protein binding via competitive displacement for binding sites on albumin (arrow denotes ↑fB). As valproic acid concentrations increase, the hepatic enzyme inhibition component of the drug interaction comes into play (arrow denotes ↓ Cl′int). The net result is total phenytoin concentrations are largely unchanged from baseline, but unbound phenytoin concentrations and pharmacologic effect increase.
INITIAL DOSAGE DETERMINATION METHODS
Several methods to initiate phenytoin therapy are available. The Pharmacokinetic Dosing method is the most flexible of the techniques. It allows individualized target serum concentrations to be chosen for a patient, and each pharmacokinetic parameter can be customized to reflect specific disease states and conditions present in the patient. Unfortunately, specific values for Michaelis-Menten pharmacokinetic variables are not known for many disease states and conditions because they are difficult to measure. Even when values are available, there is 10-15-fold variation for each parameter. Also, it is computationally intensive. Literature-based recommended dosing is a very commonly used method to prescribe initial doses of phenytoin. Doses are based on those that commonly produce steady-state concentrations in the lower end of the therapeutic range, although there is a wide variation in the actual concentrations for a specific patient.
Pharmacokinetic Dosing Method
The goal of initial dosing with phenytoin is to compute the best dose possible for the patient given their set of disease states and conditions that influence phenytoin pharmacokinetics. The optimal way to accomplish this goal is to use average parameters measured in other patients with similar disease state and condition profiles as estimates of pharmacokinetic constants for the patient currently being treated with the drug. Unfortunately, because of the difficulty in computing Michaelis-Menten parameters, accurate estimates of Vmax and Km are not available for many important patient populations. Even if average-population Michaelis-Menten constants are available, the l0-15-fold variation in these parameters means that initial doses derived from these parameters will not be successful in achieving desired goals for all patients. Phenytoin serum concentration monitoring, including unbound concentration measurement if altered plasma protein binding is suspected, is an important component of therapy for this drug. If the patient has significant hepatic dysfunction (Child-Pugh score ≥8), maintenance doses computed using this method should be decreased by 25%-50% depending on how aggressive therapy is required to be for the individual.
Michaelis-Menten Parameter Estimates
Normal adults with normal liver and renal function as well as normal plasma protein binding have an average phenytoin Vmax of 7 mg/kg/d and Km of 4 μg/mL. Michaelis-Menten parameters for younger children (6 months-6 years) are Vmax = 12 mg/kg/d and Km = 6 μg/mL while for older children (7-16 years) Vmax = 9 mg/kg/d and Km = 6 μg/mL. These are the only parameters required to estimate a maintenance dose for phenytoin.
Volume of Distribution Estimate
The volume of distribution for patients with normal phenytoin plasma protein binding is estimated at 0.7 L/kg for adults. For obese individuals 30% or more above their ideal body weight, the volume of distribution can be estimated using the following equation: V = 0.7 L/kg [IBW + 1.33(TBW − IBW)], where IBW is ideal body weight in kg [IBWfemales (in kg) = 45 + 2.3(Ht − 60) or IBWmales (in kg) = 50 + 2.3(Ht − 60)], Ht is height in inches, and TBW is total body weight in kg.74 This parameter is used to estimate the loading dose (LD in mg) for phenytoin, if one is indicated: LD = Css • V, where Css is the desired total phenytoin concentration in mg/L (note: mg/L = μg/mL and this conversion was directly made to avoid unnecessary unit conversion) and V is volume of distribution in L. For example, the volume of distribution for a 70-kg, nonobese patient would equal 49 L (V = 0.7 L/kg • 70 kg = 49 L). The loading dose to achieve a total phenytoin concentration of 15 μg/mL is 750 mg [LD = Css • V = 15 mg/L • 49 L = 735 mg, rounded to 750 mg (note: mg/L = μg/mL and this conversion was directly made to avoid unnecessary unit conversion)]. For an obese individual with a total body weight of 150 kg and an ideal body weight of 70 kg, the volume of distribution would equal 123 L: V = 0.7 L/kg [IBW + 1.33(TBW − IBW)] = 0.7 L/kg [70 kg + 1.33(150 kg − 70 kg)] = 123 L.
Selection of Appropriate Pharmacokinetic Model and Equations
When given by short-term intravenous infusion or orally, phenytoin follows a one-compartment pharmacokinetic model. When oral therapy is required, most clinicians utilize an extended phenytoin capsule dosage form that has good bioavailability (F = 1), supplies a continuous release of phenytoin into the gastrointestinal tract, and provides a smooth phenytoin serum concentration-time curve that emulates an intravenous infusion after once or twice daily dosing. Because of this, the Michaelis-Menten pharmacokinetic equation that computes the average phenytoin steady-state serum concentration (Css in μg/mL = mg/L) is widely used and allows maintenance dosage calculation:
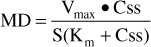
Or, solved for Css:
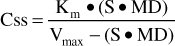
where Vmax is the maximum rate of metabolism in mg/d, S is the fraction of the phenytoin salt form that is active phenytoin (0.92 for phenytoin sodium injection and capsules; 0.92 for fosphenytoin because doses are prescribed as a phenytoin sodium equivalent or PE, 1.0 for phenytoin acid suspensions and tablets), MD is the maintenance dose of the phenytoin salt contained in the dosage form in mg/d, Css is the phenytoin concentration in mg/L (which equals μg/mL), and Km is the substrate concentration in mg/L (which equals μg/mL) where the rate of metabolism = Vmax/2.
The equation used to calculate loading doses (LD in mg) is based on a simple one-compartment model: LD = (Css • V)/S, where Css is the desired phenytoin steady-state concentration in μg/mL which is equivalent to mg/L, V is the phenytoin volume of distribution, and S is the fraction of the phenytoin salt form that is active (0.92 for phenytoin sodium injection and capsules; 0.92 for fosphenytoin because doses are prescribed as a phenytoin sodium equivalent or PE, 1.0 for phenytoin acid suspensions and tablets). Intravenous phenytoin sodium doses should be short-term infusions given no greater than 50 mg/min, and intravenous fosphenytoin doses should be short-term infusions given no greater than 150 mg/min PE.
Steady-State Concentration Selection
The general accepted therapeutic ranges for total and unbound phenytoin concentrations are 10-20 μg/mL and 1-2 μg/mL, respectively, for the treatment seizures. As previously discussed, unbound concentrations represent the portion of phenytoin that is in equilibrium with the central nervous system and should most accurately reflect drug concentration at the site of action. Thus, for patients with altered phenytoin plasma protein binding, it is more important to have the unbound concentration within its therapeutic range than the total concentration. To establish that the unbound fraction (fB) is altered for a patient, phenytoin total and unbound concentrations should be simultaneously measured from the same blood sample: fB = Cf/C, where C is the total phenytoin concentration in μg/mL and Cf is the unbound, or “free,” phenytoin concentration in μg/mL. As long as the disease states or conditions that caused altered phenytoin plasma protein binding is stable, a previously measured unbound fraction can be used to convert newly measured total phenytoin concentrations to their unbound equivalent (Cf = fBC). Phenytoin therapy must be individualized for each patient in order to achieve optimal responses and minimal side effects.
EXAMPLE 1 
TD is a 50-year-old, 75-kg (height = 5 ft 10 in) male with simple partial seizures who requires therapy with oral phenytoin. He has normal liver and renal function. Suggest an initial phenytoin dosage regimen designed to achieve a steady-state phenytoin concentration equal to 12 μg/mL.
1. Estimate Michaelis-Menten constants according to disease states and conditions present in the patient.
The Vmax for a nonobese adult patient with normal liver and renal function is 7 mg/kg/d. For a 75-kg patient, Vmax = 525 mg/d: Vmax = 7 mg/kg/d • 75 kg = 525 mg/d. For this individual, Km = 4 mg/L.
2. Compute dosage regimen.
Oral extended phenytoin sodium capsules will be prescribed to this patient (F = 1, S = 0.92). The initial dosage interval (τ) will be set to 24 hours. (Note: μg/mL = mg/L and this concentration unit was substituted for Css in the calculations so that unnecessary unit conversion was not required.) The dosage equation for phenytoin is:
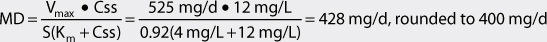
A steady-state trough total phenytoin serum concentration should be measured after steady-state is attained in 7-14 days. Phenytoin serum concentrations should also be measured if the patient experiences an exacerbation of their epilepsy, or if the patient develops potential signs or symptoms of phenytoin toxicity.
EXAMPLE 2 
UO is a 10-year-old, 40-kg male with simple partial seizures who requires therapy with oral phenytoin. He has normal liver and renal function. Suggest an initial phenytoin dosage regimen designed to achieve a steady-state phenytoin concentration equal to 12 μg/mL.
1. Estimate Michaelis-Menten constants according to disease states and conditions present in the patient.
The Vmax for a 7-16-year-old adolescent patient with normal liver and renal function is 9 mg/kg/d. For a 40-kg patient, Vmax = 360 mg/d: Vmax = 9 mg/kg/d • 40 kg = 360 mg/d. For this individual, Km = 6 mg/L.
2. Compute dosage regimen.
Oral phenytoin suspension will be prescribed to this patient (F = 1, S = 1). The initial dosage interval (τ) will be set to 12 hours. (Note: μg/mL = mg/L and this concentration unit was substituted for Css in the calculations so that unnecessary unit conversion was not required.) The dosage equation for phenytoin is:
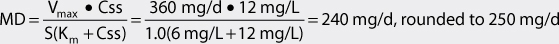
Phenytoin suspension 125 mg every 12 hours would be prescribed for the patient. A steady-state trough total phenytoin serum concentration should be measured after steady-state is attained in 7-14 days. Phenytoin serum concentrations should also be measured if the patient experiences an exacerbation of their epilepsy, or if the patient develops potential signs or symptoms of phenytoin toxicity.
To illustrate the differences and similarities between oral and intravenous phenytoin dosage regimen design, the same cases will be used to compute intravenous phenytoin or fosphenytoin loading and maintenance doses.
EXAMPLE 3 
TD is a 50-year-old, 75-kg (height = 5 ft 10 in) male with simple partial seizures who requires therapy with intravenous phenytoin sodium. He has normal liver and renal function. Suggest an initial phenytoin dosage regimen designed to achieve a steady-state phenytoin concentration equal to 12 μg/mL.
1. Estimate Michaelis-Menten and volume of distribution constants according to disease states and conditions present in the patient.
The Vmax for a nonobese adult patient with normal liver and renal function is 7 mg/kg/d. For a 75-kg patient, Vmax = 525 mg/d: Vmax = 7 mg/kg/d • 75 kg = 525 mg/d. For this individual, Km = 4 mg/L. The volume of distribution for this patient would equal 53 L: V = 0.7 L/kg • 75 kg = 53 L.
2. Compute dosage regimen.
Intravenous phenytoin sodium will be prescribed to this patient (F = 1, S = 0.92). If a loading dose is needed it would be computed using the following equation: LD = (V • Css)/S = (53 L • 12 mg/L)/0.92 = 691 mg, rounded to 700 mg given at a maximal rate of 50 mg/min. (Note: μg/mL = mg/L and this concentration unit was substituted for Css in the calculations so that unnecessary unit conversion was not required.)
For the maintenance dose, the initial dosage interval (τ) will be set to 12 hours. The dosage equation for phenytoin is:
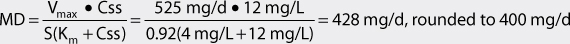
The patient would be prescribed 200 mg of phenytoin sodium injection every 12 hours using an infusion rate no greater than 50 mg/min. A steady-state trough total phenytoin serum concentration should be measured after steady-state is attained in 7-14 days. Phenytoin serum concentrations should also be measured if the patient experiences an exacerbation of their epilepsy, or if the patient develops potential signs or symptoms of phenytoin toxicity.
EXAMPLE 4 
UO is a 10-year-old, 40-kg male with simple partial seizures who requires therapy with intravenous fosphenytoin. He has normal liver and renal function. Suggest an initial phenytoin dosage regimen designed to achieve a steady-state phenytoin concentration equal to 12 μg/mL.
1. Estimate Michaelis-Menten and volume of distribution constants according to disease states and conditions present in the patient.
The Vmax for a 7-16-year-old adolescent patient with normal liver and renal function is 9 mg/kg/d. For a 40-kg patient, Vmax = 360 mg/d: Vmax = 9 mg/kg/d • 40 kg = 360 mg/d. For this individual, Km = 6 mg/L. The volume of distribution for this patient would equal 28 L: V = 0.7 L/kg • 40 kg = 28 L.
2. Compute dosage regimen.
Intravenous fosphenytoin will be prescribed, in phenytoin sodium equivalents or PE, to this patient (F = 1, S = 0.92). If a loading dose is needed, it would be computed using the following equation: LD = (V • Css)/ S = (28 L • 12 mg/L)/0.92 = 365 mg, rounded to 350 mg given at a maximal rate of 150 mg/min PE. (Note: μg/mL = mg/L and this concentration unit was substituted for Css in the calculations so that unnecessary unit conversion was not required.) The dosage equation for phenytoin is:
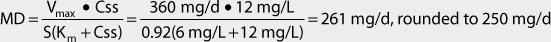
Intravenous fosphenytoin 125 mg PE every 12 hours given no greater than 150 mg/min PE would be prescribed for the patient. A steady-state trough total phenytoin serum concentration should be measured after steady-state is attained in 7-14 days. Phenytoin serum concentrations should also be measured if the patient experiences an exacerbation of their epilepsy, or if the patient develops potential signs or symptoms of phenytoin toxicity.
LITERATURE-BASED RECOMMENDED DOSING
Because of the large amount of variability in phenytoin pharmacokinetics, even when concurrent disease states and conditions are identified, many clinicians believe that the use of standard phenytoin doses for various situations are warranted. The original computations of these doses were based on the Pharmacokinetic Dosing methods described in the previous section, and subsequently modified based on clinical experience. In general, the expected phenytoin steady-state serum concentrations used to compute these doses was 10-15 μg/mL. Suggested phenytoin maintenance doses are 4-6 mg/kg/d for adults and 5-10 mg/kg/d for children (6 months-16 years old). Phenytoin-loading doses are 15-20 mg/kg. For obese individuals (>30% over ideal body weight), adjusted body weight (ABW) should be used to compute loading doses: ABW (in kg) = IBW + 1.33(TBW − IBW), where IBW is ideal body weight in kg [IBWfemales (in kg) = 45 + 2.3(Ht − 60) or IBWmales (in kg) = 50 + 2.3(Ht − 60)], Ht is height in inches, and TBW is total body weight in kg.74 Although clearance probably is increased in obese individuals, precise information regarding the best weight factor is lacking for maintenance dose computation, so most clinicians use ideal body weight to calculate this dose. If the patient has significant hepatic dysfunction (Child-Pugh score ≥8), maintenance doses prescribed using this method should be decreased by 25%-50% depending on how aggressive therapy is required to be for the individual. Doses of phenytoin, phenytoin sodium, or fosphenytoin (in PE or phenytoin sodium equivalents) are computed using these dosage rates since dosage amounts will be rounded to clinically acceptable amounts.
To illustrate the similarities and differences between this method of dosage calculation and the Pharmacokinetic Dosing method, the same examples used in the previous section will be used.
EXAMPLE 1 
TD is a 50-year-old, 75-kg (height = 5 ft 10 in) male with simple partial seizures who requires therapy with oral phenytoin. He has normal liver and renal function. Suggest an initial phenytoin dosage regimen designed to achieve a steady-state phenytoin concentration equal to 12 μg/mL.
1. Estimate phenytoin dose according to disease states and conditions present in the patient.
The suggested initial dosage rate for extended phenytoin sodium capsules in an adult patient is 4-6 mg/kg/d. Using a rate of 5 mg/kg/d, the initial dose would be 400 mg/d: 5 mg/kg/d • 75 kg = 375 mg/d, rounded to 400 mg/d. Using a dosage interval of 24 hours, the prescribed dose would be 400 mg of extended phenytoin sodium capsules daily.
A steady-state trough total phenytoin serum concentration should be measured after steady-state is attained in 7-14 days. Phenytoin serum concentrations should also be measured if the patient experiences an exacerbation of their epilepsy, or if the patient develops potential signs or symptoms of phenytoin toxicity.
EXAMPLE 2 
UO is a 10-year-old, 40-kg male with simple partial seizures who requires therapy with oral phenytoin. He has normal liver and renal function. Suggest an initial phenytoin dosage regimen designed to achieve a steady-state phenytoin concentration equal to 12 μg/mL.
1. Estimate phenytoin dose according to disease states and conditions present in the patient.
The suggested initial dosage rate for phenytoin suspension in an adolescent patient is 5-10 mg/kg/d. Using a rate of 6 mg/kg/d, the initial dose would be 250 mg/d: 6 mg/kg/d • 40 kg = 240 mg/d, rounded to 250 mg/d. Using a dosage interval of 12 hours, the prescribed dose would be 125 mg of phenytoin suspension every 12 hours.
A steady-state trough total phenytoin serum concentration should be measured after steady-state is attained in 7-14 days. Phenytoin serum concentrations should also be measured if the patient experiences an exacerbation of their epilepsy, or if the patient develops potential signs or symptoms of phenytoin toxicity.
To illustrate the differences and similarities between oral and intravenous phenytoin dosage regimen design, the same cases will be used to compute intravenous phenytoin or fosphenytoin loading and maintenance doses.
EXAMPLE 3 
TD is a 50-year-old, 75-kg (height = 5 ft 10 in) male with simple partial seizures who requires therapy with intravenous phenytoin sodium. He has normal liver and renal function. Suggest an initial phenytoin dosage regimen designed to achieve a steady-state phenytoin concentration equal to 12 μg/mL.
1. Estimate phenytoin dose according to disease states and conditions present in the patient.
The suggested initial dosage rate for phenytoin sodium injection in an adult patient is 4-6 mg/kg/d. Using a rate of 5 mg/kg/d, the initial dose would be 400 mg/d: 5 mg/kg/d • 75 kg = 375 mg/d, rounded to 400 mg/d. Using a dosage interval of 12 hours, the prescribed dose would be 200 mg of phenytoin sodium injection every 12 hours. If loading dose administration was necessary, the suggested amount is 15-20 mg/kg. Using 15 mg/kg, the suggested loading dose would be 1250 mg of phenytoin sodium injection given no faster than 50 mg/min: 15 mg/kg • 75 kg = 1125 mg, rounded to 1250 mg.
A steady-state trough total phenytoin serum concentration should be measured after steady-state is attained in 7-14 days. Phenytoin serum concentrations should also be measured if the patient experiences an exacerbation of their epilepsy, or if the patient develops potential signs or symptoms of phenytoin toxicity.
EXAMPLE 4 
UO is a 10-year-old, 40-kg male with simple partial seizures who requires therapy with intravenous fosphenytoin. He has normal liver and renal function. Suggest an initial phenytoin dosage regimen designed to achieve a steady-state phenytoin concentration equal to 12 μg/mL.
1. Estimate phenytoin dose according to disease states and conditions present in the patient.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


