FIGURE 6-1 Digoxin serum concentrations after 250-μg doses given intravenously (circles and solid line) and orally as a tablet (squares with dashed line). After an intravenous dose, digoxin serum concentrations are very high because the entire quantity of drug is initially contained in the blood. During the distribution phase, digoxin begins to move out of the vascular system into the tissues. It is also cleared from the body during this phase. Digoxin serum concentrations decline relatively rapidly over an 8-12-hour time period until the blood and tissues are in pseudoequilibrium with each other. During the elimination phase, digoxin serum concentrations in patients with good renal function (creatinine clearance > 80 mL/min) decline with a half-life of about 36 hours. After oral tablet administration, about 70% of a digoxin dose is absorbed from the gastrointestinal tract. Maximum, or peak, concentrations occur about 1.5-2 hours after oral dosing with tablets, and the distribution phase still lasts 8-12 hours. During the elimination phase, intravenous and oral digoxin have the same terminal half-lives.
There is a great deal of inter- and intrapatient variability in the pharmacodynamic responses to digoxin. Clinically beneficial inotropic effects of digoxin are generally achieved at steady-state serum concentrations of 0.5-1 ng/mL.13,14 Increasing steady-state serum concentrations to 1.2-1.5 ng/mL may provide some minor, additional inotropic effect.13,14 Chronotropic effects usually require higher digoxin steady-state serum concentrations of 0.8-1.5 ng/mL.15,16 Additional chronotropic effects may be observed at digoxin steady-state serum concentrations as high as 2 ng/mL. Because of pharmacodynamic variability, clinicians should consider these ranges as initial guidelines and rely heavily on patient response to monitor digoxin therapy.
Steady-state digoxin serum concentrations above 2 ng/mL are associated with an increased incidence of adverse drug reactions. At digoxin concentrations of 2.5 ng/mL or above ~50% of all patients will exhibit some form of digoxin toxicity.17 Most digoxin side effects involve the gastrointestinal tract, central nervous system, or cardiovascular system.18 Gastrointestinal tract–related adverse effects include anorexia, nausea, vomiting, diarrhea, abdominal pain, or constipation. Central nervous system side effects are headache, fatigue, insomnia, confusion, or vertigo. Visual disturbances can also occur and are manifested as blurred vision and changes in color vision or colored halos around objects oftentimes involving the yellow-green spectrum. As can be appreciated, most of the gastrointestinal and central nervous system side effects of digoxin are nonspecific and could be caused by many different things. Because of this, clinicians should pay close attention to any new symptoms reported by patients receiving cardiac glycosides. Cardiac side effects commonly include second- or third-degree atrioventricular block, atrioventricular dissociation, bradycardia, premature ventricular contractions, or ventricular tachycardia. Rarely, almost every cardiac arrhythmia has been reported to occur due to digoxin toxicity. If a patient develops a new arrhythmia while receiving digoxin treatment, consideration should be given to the possibility that it is digoxin induced. Also, it should be noted that relatively minor adverse effects such as nausea, headache, or changes in color vision may not occur in a patient before major cardiovascular side effects are found. In the case of life-threatening digoxin overdose, digoxin antigen-binding fragments or digoxin immune FAB (DigiFab) are portions of digoxin-specific antibodies that can be used to rapidly reverse the adverse symptoms (see Special Dosing Considerations section).
CLINICAL MONITORING PARAMETERS
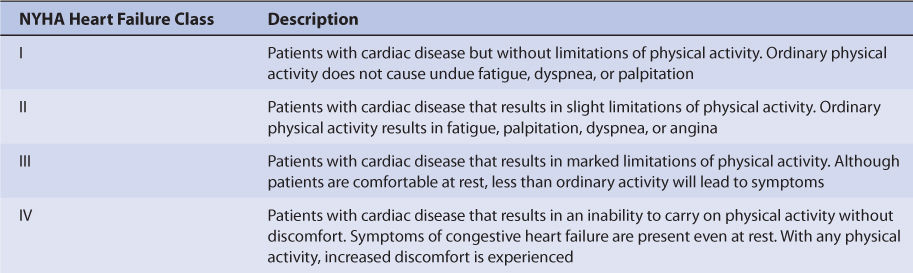
In patients receiving digoxin for heart failure, the common signs and symptoms of CHF should be routinely monitored—left-sided failure: dyspnea on exertion, paroxysmal nocturnal dyspnea, orthopnea, tachypnea, cough, hemoptysis, pulmonary rales/edema, S3 gallop, pleural effusion, Cheyne-Stokes respiration; right-sided failure: abdominal pain, anorexia, nausea, bloating, constipation, ascites, peripheral edema, jugular venous distention, hepatojugular reflux, hepatomegaly; general symptoms: fatigue, weakness, nocturia, CNS symptoms, tachycardia, pallor, digital cyanosis, cardiomegaly.6 A very useful functional classification for heart failure patients proposed by the New York Heart Association is given in Table 6-1.
When used for the treatment of atrial fibrillation, digoxin will not stop the atrial arrhythmia but is used to decrease, or control, the ventricular rate to an acceptable value (usually <100 beats/min).1 The patient’s pulse or ventricular rate should be monitored, and an electrocardiogram can also be useful to clinicians able to interpret the output. Atrial fibrillation is characterized by 400-600 nonuniform atrial beats/min. Sinus rhythm will not be restored with the use of digoxin alone although atrial fibrillation can spontaneously remit. Depending on the symptomatology experienced by the patient, cardioversion can be attempted by using direct electrical current or by the use of an antiarrhythmic agent such as flecainide, dofetilide, propafenone, or ibutilide.4 Adequate anticoagulation to prevent thromboembolism is needed before cardioversion if atrial fibrillation has occurred for longer than 48 hours or the duration of atrial fibrillation is unknown. Optionally, a transesophageal echocardiogram (TEE) can be performed, and if no thrombus is identified in the left atrium or left atrium appendage, cardioversion can commence after adequate anticoagulation with unfractionated heparin has been established.4
Patients with severe heart disease such as coronary artery disease (angina, myocardial infarction) can have increased pharmacodynamic sensitivity to cardiac glycosides, and patients receiving these drugs should be monitored closely for adverse drug effects.17,19 Also, augmented pharmacologic responses to digitalis derivatives occur with serum electrolyte disturbances such as hypokalemia, hypomagnesemia, and hypercalcemia even though steady-state digoxin serum concentrations are in the therapeutic range.7,9 Serum potassium concentrations should be routinely monitored in patients receiving digoxin and potassium-wasting diuretics. Potassium supplementation may be necessary in some of these patients. Also, many patients receiving digoxin and diuretics will be receiving angiotensin I converting enzyme (ACE) inhibitors or angiotensin II receptor blockers (ARB) which can cause potassium retention. When receiving a combination of these drugs, it can be difficult to reasonably ascertain what the patient’s serum potassium status is without measuring it.
As an adjunct to the patient’s clinical response, post-distribution (8-12 hours post-dose) steady-state digoxin serum concentrations can be measured 3-5 half-lives after a stable dose is initiated. Digoxin is primarily eliminated unchanged by the kidney (~75%), so its clearance is predominately influenced by renal function.11,12 Once stable, therapeutic steady-state digoxin serum concentrations and dosage levels have been established, serum creatinine measurements can be used to detect changes in renal function which may result in digoxin clearance and concentration alterations. Hospitalized patients with severe or acute heart failure may need to have serum creatinine determinations two or three times weekly to monitor renal function, while ambulatory patients with stable heart failure may only need yearly serum creatinine measurements.
BASIC CLINICAL PHARMACOKINETIC PARAMETERS
The primary route of digoxin elimination from the body is by the kidney via glomerular filtration and active tubular secretion of unchanged drug (~75%).11,12 The remainder of a digoxin dose (~25%) is removed by hepatic metabolism or biliary excretion. The primary transporter involved in active tubular secretion and biliary excretion is P-glycoprotein (PGP).20,21 Enterohepatic recirculation (reabsorption of drug from the gastrointestinal tract after elimination in the bile) of digoxin occurs.22 Digoxin is given as an intravenous injection or orally as a tablet or solution. When given intravenously, doses should be infused over at least 5-10 minutes. Average bioavailability constants (F) for the tablet and oral solution are 0.7 and 0.8.23–28 Digoxin is not usually administered intramuscularly due to erratic absorption and severe pain at the injection site. Plasma protein binding is ~25% for digoxin.29,30 Usual digoxin doses for adults are 250 μg/d (range: 125-500 μg/d) in patients with good renal function (creatinine clearance ≥ 80 mL/min) and 125 μg every 2-3 days in patients with renal dysfunction (creatinine clearance ≤ 15 mL/min).
EFFECTS OF DISEASE STATES AND CONDITIONS ON DIGOXIN PHARMACOKINETICS AND DOSING
Adults with normal renal function (creatinine clearance ≥ 80 mL/min, Table 6-2) have an average digoxin half-life of 36 hours (range: 24-48 hours) and volume of distribution of 7 L/kg (range: 5-9 L/kg).31,32 The volume of distribution is large due to the extensive tissue binding of digoxin in the body. Digoxin pharmacokinetics are not affected by obesity (>30% over ideal body weight), so volume of distribution and dosage estimates should be based on ideal body weight.33,34
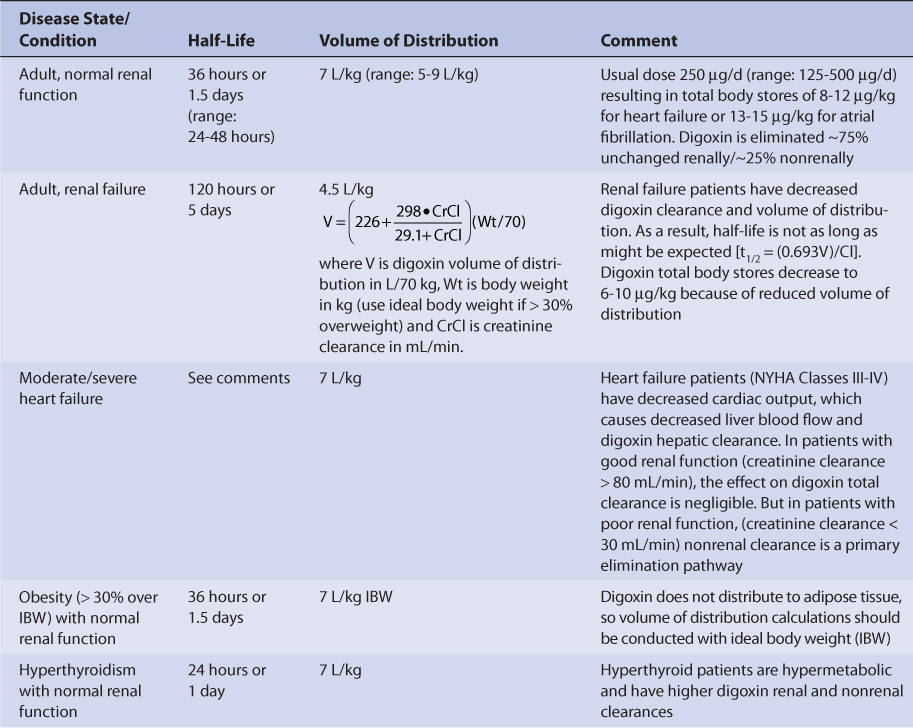
Because digoxin is principally eliminated by the kidney, renal dysfunction is the most important disease state that effects digoxin pharmacokinetics.12 The digoxin clearance rate decreases in proportion to creatinine clearance, and this relationship will be utilized to aid in the computation of initial doses later in the chapter (Figure 6-2). The equation that estimates digoxin clearance from creatinine clearance is: Cl = 1.303 (CrCl) + ClNR, where Cl is digoxin clearance in mL/min, CrCl is creatinine clearance in mL/min, and ClNR is digoxin clearance by nonrenal routes of elimination, which equals 40 mL/min in patients with no or mild heart failure (NYHA CHF class I or II, see Table 6-1).12 Digoxin volume of distribution, in addition to clearance, decreases with declining renal function.10,35 While the mechanism for this change is not as well understood, it is likely that digoxin is displaced from tissue-binding sites by an unknown substance or substances present in patients with renal dysfunction so that drug which would have been bound to tissues becomes unbound. Unbound digoxin molecules displaced from tissue-binding sites move into the blood causing the decreased volume of distribution [↓V = Vb + (fb/↑ft) Vt, where V is digoxin volume of distribution, Vb is blood volume, Vt is tissue volume, fb is the unbound fraction of digoxin in the blood, and ft is the unbound fraction of digoxin in the tissues]. The equation that estimates digoxin volume of distribution using creatinine clearance is:
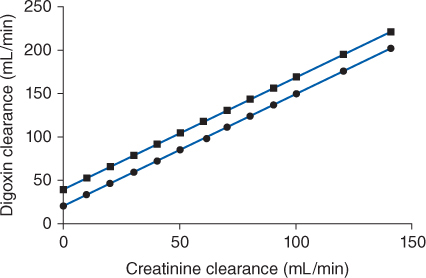
FIGURE 6-2 Digoxin clearance is proportional to creatinine clearance for patients with [circles with solid line: Cl = 1.303(CrCl) + 20] and without [squares with dashed line: Cl = 1.303(CrCl) + 40] moderate-severe (NYHA Class III or IV) heart failure. Nonrenal clearance (denoted by the y-intercept) is lower for patients with moderate-severe heart failure because reduced cardiac output results in decreased liver blood flow and digoxin hepatic clearance.
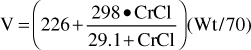
where V is digoxin volume of distribution in L/70 kg, Wt is body weight in kg (use ideal body weight if >30% overweight), and CrCl is creatinine clearance in mL/min.35 Because digoxin volume of distribution and clearance decrease simultaneously in patients with renal failure, the average half-life for digoxin of 5 days is shorter than what might be expected if clearance alone decreased [t1/2 = (0.693 • V)/Cl].
Digoxin is not significantly eliminated by hemodialysis or peritoneal dialysis.31,32 Hemofiltration does remove digoxin with a typical sieving coefficient of 0.7.36,37 In many cases, a sufficient amount of digoxin will not be removed to warrant an increased maintenance dose. However, due to pharmacokinetic variability, some patients may need a periodic booster dose to increase digoxin concentrations (see Special Dosing Consideration section at end of chapter).37
Heart failure decreases cardiac output, which in turn decreases liver blood flow. Liver blood flow is an important factor in the determination of hepatic clearance for drugs because it is the vehicle that delivers drug molecules to the liver for possible elimination. Moderate-severe heart failure (NYHA CHF class III or IV, see Table 6-1) decreases the hepatic clearance of digoxin by this mechanism.12 When estimating digoxin clearance for the purpose of computing initial drug doses, it is necessary to decrease the nonrenal clearance (ClNR) factor to 20 mL/min in the equation to compensate for decreased hepatic clearance: Cl = 1.303 (CrCl) + 20, where Cl is digoxin clearance in mL/min, CrCl is creatinine clearance in mL/min, and 20 is digoxin nonrenal clearance ClNR in mL/min.
Thyroid hormone regulates basil metabolic rate, and thyroid status will influence every major organ system in the body including the heart (heart rate and cardiac output), liver (liver blood flow and microsomal drug-metabolizing enzyme function), and kidney (renal blood flow and glomerular filtration rate). Patients who are hypothyroid will have slower metabolic rates and eliminate digoxin more slowly than euthyroid patients (t1/2 = 48 hours with normal renal function).31,32,38–40 Hyperthyroid patients have faster metabolic rates and eliminate digoxin faster than euthyroid patients (t1/2 = 24 hours with normal renal function).31,32,38–40 Hyperthyroid patients can present with atrial fibrillation which may be treated with digoxin. Generally, these patients require higher digoxin doses to control ventricular rate because of the increase in digoxin clearance.
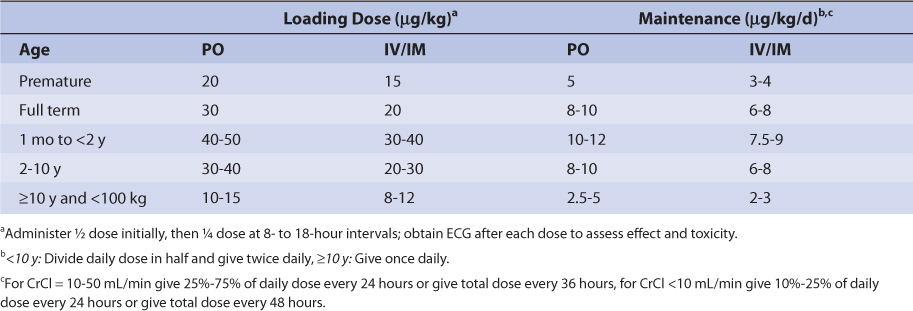
Similar to other drugs, digoxin clearance is lower in neonates and premature infants because renal and hepatic function are not completely developed.41,42 Premature infants and neonates have average digoxin half-lives equal to 60 hours and 45 hours, respectively. In older babies and young children (6 months-8 years old), renal and hepatic function are fully developed and half-lives can be as short as 18 hours. Older children (≥ 12 years old) have mean digoxin half-lives (t1/2 = 36 hours) that are similar to those found in adults. Also, volume of distribution is larger in infants and children compared to adults as is found with many other drugs. Pediatric loading and maintenance doses are given in Table 6-3.
TABLE 6-3 Initial Pediatric Doses of Digoxin for Patients With Normal Renal Function (CrCl > 50 mL/min)98
Malabsorption of oral digoxin has been reported in patients with severe diarrhea, radiation treatments to the abdomen, and gastrointestinal hypermotility.38,43–47 In these cases, steady-state digoxin serum concentrations decrease due to poor bioavailability of the drug.
DRUG INTERACTIONS
Digoxin has an extensive list of drug interactions with other agents. Because of this, only the most common and severe drug interactions will be discussed. Inhibition of P-glycoprotein, a drug efflux pump which is found in the kidney, liver, and intestine, appears to be involved in the majority of digoxin interactions.20,21,48 Clinicians should consult a current drug interaction reference when other medications are prescribed to patients receiving digoxin therapy.49
Quinidine decreases both the renal and nonrenal clearance of digoxin and also decreases the volume of distribution of digoxin.50–55 Inhibition of P-glycoprotein may be involved in this interaction.48 The result of this complex interaction is that concurrent quinidine therapy increases the average steady-state digoxin concentration by 30%-70%.
Verapamil, diltiazem, and bepridil inhibit digoxin clearance and increase mean digoxin steady-state concentrations by various degrees.55–61 Of these calcium channel blockers, verapamil is the most potent inhibitor of digoxin clearance, and increases digoxin steady-state serum concentrations up to 70%. Diltiazem and bepridil therapy each increase average digoxin steady-state serum concentrations by about 30%.
Amiodarone62–65 and propafenone66–68 are antiarrhythmic agents that decrease digoxin clearance. In addition to this drug interaction mechanism, amiodarone also simultaneously increases digoxin oral bioavailability, and it is likely that P-glycoprotein inhibition is involved in the drug interaction between these two drugs.69 Digoxin steady-state serum concentrations increase two-three times over baseline values with concomitant amiodarone therapy. Because amiodarone has a very long half-life (~50 hours), the onset of the drug interaction with digoxin can be very long. As serum concentrations of amiodarone slowly increase and approach steady-state values, digoxin clearance and bioavailability are simultaneously slowly changing. The insidious nature of the amiodarone–digoxin drug interaction can make it difficult to detect in patients. Propafenone therapy increases mean digoxin steady-state concentrations by 30%-60% in a dose-dependent fashion with propafenone doses of 450 mg/d causing digoxin concentration changes in the lower end of the range and propafenone doses of 900 mg/d causing digoxin concentration changes in the upper end of the range.
Cyclosporine therapy has been reported to increase average steady-state digoxin concentrations up to 50%.70 P-glycoprotein inhibition by cyclosporine is the primary mechanism for this drug interaction.20
About 10% of patients receiving digoxin therapy have significant amounts of Eubacterium lentum in their gastrointestinal tract that metabolizes orally administered digoxin before it can be absorbed.71,72 Erythromycin, clarithromycin, and tetracycline are antibiotics that can kill this bacteria.73–78 Digoxin steady-state serum concentrations increase an average of 30% in these select patients when one of these three antibiotics have been prescribed. P-glycoprotein inhibition may be one of the mechanisms involved with this interaction involving macrolide antibiotics.78
The absorption of oral digoxin from the gastrointestinal tract is influenced by many different compounds. Aluminum-containing antacids and kaolin-pectin physically adsorb digoxin, rendering it unabsorbable.79 These compounds should be administered no closer than 2 hours to an oral digoxin dose. Similarly, cholestyramine also reduces digoxin oral bioavailability by binding it in the gastrointestinal tract and should be given no closer than 8 hours to a digoxin oral dose.80,81 Sulfasalazine and neomycin each decrease digoxin oral bioavailability by unknown mechanisms.82,83 Propantheline increases oral digoxin bioavailability by prolonging gastrointestinal transit time, while metoclopramide and cisapride decreases oral digoxin bioavailability by decreasing gastrointestinal transit time.81,84,85
INITIAL DOSAGE DETERMINATION METHODS
Several methods to initiate digoxin therapy are available. The Pharmacokinetic Dosing method is the most flexible of the techniques. It allows individualized target serum concentrations to be chosen for a patient, and each pharmacokinetic parameter can be customized to reflect specific disease states and conditions present in the patient. However, it is computationally intensive.
The Jelliffe method is similar to the Pharmacokinetic Dosing method, except a target total body store is selected based on specific disease states and conditions present in the patient. It is also computationally intensive.
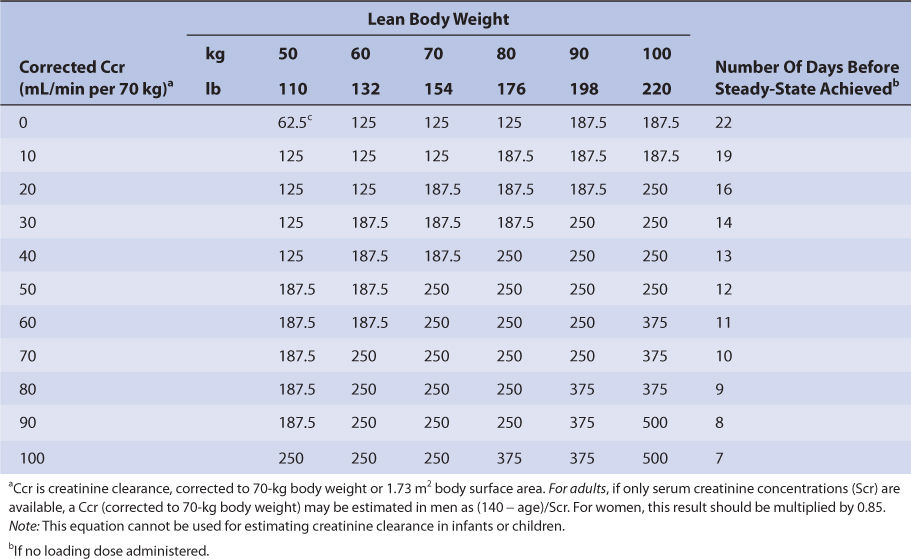
Nomograms that use the dosing concepts in the Jelliffe dosing method are available. But, in order to make calculations easier, they make simplifying assumptions. The nomograms are for adults only, and separate versions are needed for intravenous injection (Table 6-4A) and tablets (Table 6-4B) because of bioavailability differences among dosage forms. Both nomograms assume that digoxin total body stores of 10 μg/kg are adequate, so they are limited to heart failure patients requiring this dose. Another nomogram that makes other simplifying assumptions is also available for heart failure patients.86
TABLE 6-4A Jelliffe Nomogram for Intravenous Digoxin (in μg) in Adult Patients With Heart Failure to Provide Total Body Stores of 10 μg/kg99
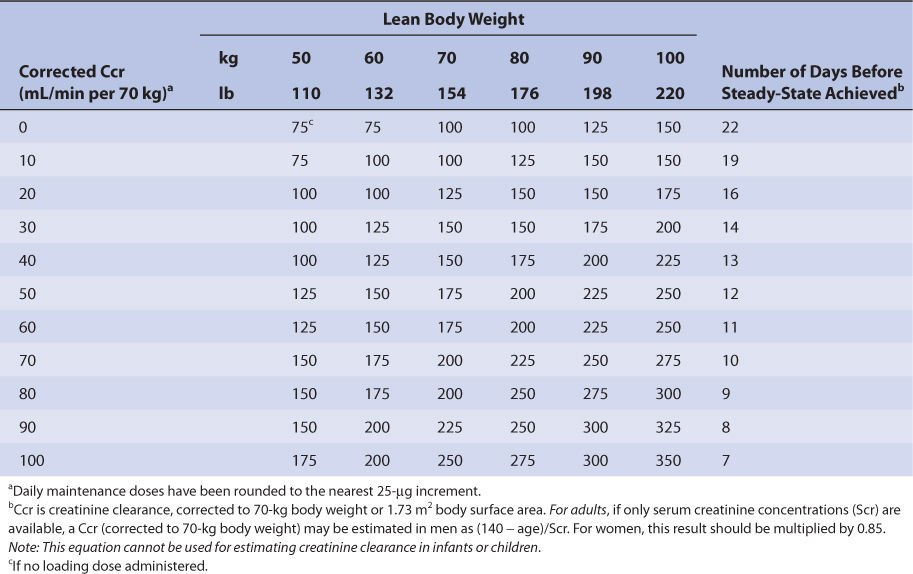
TABLE 6-4B Jelliffe Nomogram for Oral Digoxin Tablets (in μg) in Adult Patients With Heart Failure to Provide Total Body Stores of 10 μg/kg99
Recommended initial doses for pediatric patients are given in Table 6-3.
PHARMACOKINETIC DOSING METHOD
The goal of initial dosing of digoxin is to compute the best dose possible for the patient given their set of disease states and conditions that influence digoxin pharmacokinetics and the cardiovascular disorder being treated. In order to do this, pharmacokinetic parameters for the patient will be estimated using average parameters measured in other patients with similar disease state and condition profiles. This approach is also known as the Jusko-Koup method for digoxin dosing.12,35
Clearance Estimate
Digoxin is predominately eliminated unchanged in the urine, and there is a good relationship between creatinine clearance and digoxin clearance (see Figure 6-2). This relationship allows the estimation of the digoxin clearance for a patient which can be used to compute an initial dose of the cardiac glycoside. Mathematically, the equation for the straight line shown in Figure 6-2 is: Cl = 1.303 (CrCl) + ClNR, where Cl is the digoxin clearance in mL/min, CrCl is creatinine clearance in mL/min, and ClNR is digoxin nonrenal clearance.12 A digoxin nonrenal clearance value of 40 mL/min is used for patients without heart failure or who have only mild signs and symptoms of heart failure (NYHA CHF classes I or II). Patients with moderate or severe heart failure (NYHA CHF classes III or IV) have significant decreases in cardiac output which leads to a reduction in liver blood flow and digoxin hepatic clearance. In these cases, digoxin nonrenal clearance is set to equal 20 mL/min in the equation. For example, the estimated digoxin clearance for an individual with a creatinine clearance of 10 mL/min is 53 mL/min if the patient has no symptoms or mild symptoms of heart failure [Cl = 1.303 (10 mL/min) + 40 = 53 mL/min], or 33 mL/min if the patient has moderate-to-severe symptoms of heart failure [Cl = 1.303 (10 mL/min) + 20 = 33 mL/min]. Taking the patient’s renal function into account when deriving initial doses of digoxin is the single most important characteristic to assess.
Volume of Distribution Estimate
The average volume of distribution for patients without disease states and conditions that change this parameter is 7 L/kg.31,32 Because obesity does not change digoxin volume of distribution, the weight factor used in this calculation is ideal body weight (IBW) for patients that are significantly overweight (>30% over IBW).33,34 Thus, for a 70-kg patient with good renal function, the estimated volume of distribution would be 490 L (V = 7 L/kg • 70 kg = 490 L). If a patient weighs less than their ideal body weight, actual body weight is used to estimate volume of distribution. For patients whose weight is between their ideal body weight and 30% over ideal weight, actual body weight can be used to compute estimated volume of distribution, although some clinicians prefer to use ideal body weight for these individuals. In patients who are more than 30% above their ideal body weight, volume of distribution (V) estimates should be based on ideal body weight. For an obese patient with normal renal function whose ideal body weight is 55 kg and total body weight is 95 kg, the estimated volume of distribution would be 385 L: V = 7 L/kg • IBW = 7 L/kg (55 kg) = 385 L.
For patients with renal dysfunction (creatinine clearance ≤ 30 mL/min), creatinine clearance should be used to provide an improved volume of distribution estimate (V in L) using the following formula:
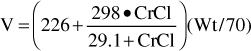
where CrCl is the patient’s creatinine clearance in mL/min.35 For example, a 70-kg patient with significant renal dysfunction (CrCl = 10 mL/min) is to receive a loading dose of digoxin and an estimate of digoxin volume of distribution is needed. The estimated volume of distribution for this patient would be 302 L:
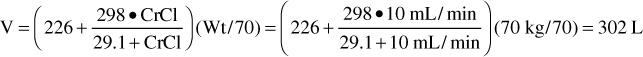
In patients who are more than 30% above their ideal body weight, volume of distribution (V) estimates should be based on ideal body weight, so the weight factor used in the equation would be IBW.
Selection of Appropriate Pharmacokinetic Model and Equations
When given by intravenous injection or orally, digoxin follows a two-compartment pharmacokinetic model (see Figure 6-1). After the end of intravenous infusion or after peak concentration has been reached after an oral dose, serum concentrations drop over an 8-12-hour time period because of distribution of drug from blood to tissues (α or distribution phase). After distribution of digoxin is complete, drug concentrations decline more slowly, and the elimination rate constant for this segment of the concentration/time curve is the one that varies with renal function (β or elimination phase). While this model is the most correct from a strict pharmacokinetic viewpoint, it cannot easily be used clinically because of its mathematical complexity. During the elimination phase of the concentration/time curve, digoxin serum concentrations drop very slowly due to the long elimination half-life (36 hours with normal renal function, 5 days with end-stage renal disease). Because of this, a very simple pharmacokinetic equation that computes the average digoxin steady-state serum concentration (Css in ng/mL = μg/L) is widely used and allows maintenance dosage calculation: Css = [F (D/τ)]/Cl or D/τ = (Css • Cl)/F, where F is the bioavailability fraction for the oral dosage form (F = 1 for intravenous digoxin), D is the digoxin dose in μg, τ is the dosage interval in days, and Cl is digoxin clearance in L/d.12,35
The equation used to calculate loading dose (LD in μg) is based on a simple one-compartment model: LD = (Css • V)/F, where Css is the desired digoxin steady-state concentration in μg/L which is equivalent to ng/mL, V is the digoxin volume of distribution, and F is the bioavailability fraction for the oral dosage form (F = 1 for intravenous digoxin). When digoxin loading doses are administered, they are usually given in divided doses separated by 4-6 hours (50% of dose at first, followed by two additional doses of 25%). A portion of the loading dose can be withheld if the patient is experiencing any digoxin adverse effects such as a low pulse rate. This technique is used to allow the assessment of clinical response before additional digoxin is given in order to avoid accidental overdosage.
Steady-State Concentration Selection
Digoxin steady-state concentrations are selected based on the cardiovascular disease being treated. For heart failure, steady-state serum concentrations of 0.5-1 ng/mL are usually effective.13,14 For initial dosing purposes, a target digoxin concentration equal to 0.8 ng/mL is reasonable. For patients with atrial fibrillation, steady-state serum concentrations of 0.8-1.5 ng/mL are usually needed to control the ventricular rate to 100 beats/min or less.15,32 An initial target digoxin concentration of 1.2 ng/mL is reasonable for patients with this disease state.
EXAMPLE 1 
MJ is a 50-year-old, 70-kg (height = 5 ft 10 in) male with atrial fibrillation for less than 24 hours. His current serum creatinine is 0.9 mg/dL, and it has been stable over the last 5 days since admission. Compute an intravenous digoxin dose for this patient to control ventricular rate.
1. Estimate creatinine clearance.
This patient has a stable serum creatinine and is not obese. The Cockcroft-Gault equation can be used to estimate creatinine clearance:
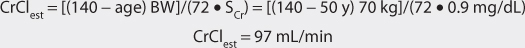
2. Estimate clearance.
The drug clearance versus creatinine clearance relationship is used to estimate the digoxin clearance for this patient (ClNR = 40 mL/min since the patient does not have moderate-severe heart failure):

3. Use average steady-state concentration equation to compute digoxin maintenance dose.
For a patient with atrial fibrillation, the desired digoxin concentration would be 0.8-1.5 ng/mL. A serum concentration equal to 1.2 ng/mL will be chosen for this patient, and intravenous digoxin will be used (F = 1). Note that for concentration units ng/mL = μg/L, and this conversion will be made before the equation is used. Also, conversion factors are needed to change milliliter units to liter (1000 mL/L) and minute units to days (1440 min/d).

4. Use loading dose equation to compute digoxin loading dose (if needed).
The patient has good renal function and is nonobese. Therefore, a volume of distribution equal to 7 L/kg and actual body weight can be used to compute the digoxin loading dose. An intravenous loading dose (F = 1) could be used in this patient to achieve the desired pharmacologic effect quicker than would occur if maintenance doses alone were used and concentrations allowed to accumulate over 3-5 half-lives.

When digoxin loading doses are administered, they are usually given in divided doses separated by 4-6 hours (50% of dose at first, followed by two additional doses of 25%). In this case, an initial intravenous dose of 250 μg would be given initially, followed by two additional intravenous doses of 125 μg each. One of the loading doses could be withheld if pulse rate was less than 50-60 beats/min or other undesirable digoxin adverse effects were noted.
EXAMPLE 2 
Same patient profile as in example 1, but serum creatinine is 3.5 mg/dL indicating renal impairment.
1. Estimate creatinine clearance.
This patient has a stable serum creatinine and is not obese. The Cockcroft-Gault equation can be used to estimate creatinine clearance:
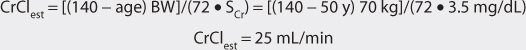
2. Estimate clearance.
The drug clearance versus creatinine clearance relationship is used to estimate the digoxin clearance for this patient (ClNR = 40 mL/min since the patient does not have moderate-severe heart failure):

3. Use average steady-state concentration equation to compute digoxin maintenance dose.
For a patient with atrial fibrillation, the desired digoxin concentration would be 0.8-1.5 ng/mL. A serum concentration equal to 1.2 ng/mL will be chosen for this patient, and intravenous digoxin will be used (F = 1). Note that for concentration units ng/mL = μg/L, and this conversion will be made before the equation is used. Also, conversion factors are needed to change milliliter units to liter (1000 mL/L) and minute units to days (1440 min/d).

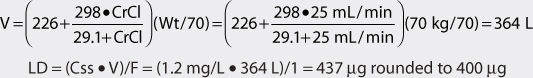
4. Use loading dose equation to compute digoxin loading dose (if needed).
The patient has poor renal function and is nonobese. Therefore, the volume of distribution equation that adjusts the parameter estimate for renal dysfunction can be used to compute the digoxin loading dose. An intravenous loading dose (F = 1) could be given in this patient to achieve the desired pharmacologic effect quicker than would occur if maintenance doses alone were used to allow concentrations to accumulate over 3-5 half-lives.
When digoxin loading doses are administered, they are usually given in divided doses separated by 4-6 hours (50% of dose at first, followed by two additional doses of 25%). In this case, an initial intravenous dose of 200 μg would be given initially, followed by two additional intravenous doses of 100 μg each. One of the loading doses could be withheld if the pulse rate was less than 50-60 beats/min or other undesirable digoxin adverse effects were noted.

Same patient profile as in example 1, but serum creatinine is 3.5 mg/dL indicating renal impairment. Additionally, the patient is being treated for NYHA Class III moderate heart failure, not atrial fibrillation. Compute an oral digoxin tablet maintenance dose for this patient.
1. Estimate creatinine clearance.
This patient has a stable serum creatinine and is not obese. The Cockcroft-Gault equation can be used to estimate creatinine clearance:
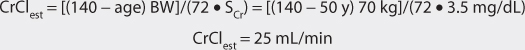
2. Estimate clearance.
The drug clearance versus creatinine clearance relationship is used to estimate the digoxin clearance for this patient (ClNR = 20 mL/min since the patient has moderate heart failure):

3. Use average steady-state concentration equation to compute digoxin maintenance dose.
For a patient with heart failure, the desired digoxin concentration would be 0.5-1 ng/mL. A serum concentration equal to 0.8 ng/mL will be chosen for this patient, and oral digoxin will be used (F = 0.7). Note that for concentration units ng/mL = μg/L, and this conversion will be made before the equation is used. Also, conversion factors are needed to change milliliter units to liter (1000 mL/L) and minute units to days (1440 min/d).

This oral tablet dose would be rounded to 125 μg every other day.
EXAMPLE 4 
OI is a 65-year-old, 170-kg (height = 5 ft 5 in) female with NYHA Class III moderate heart failure. Her current serum creatinine is 4.7 mg/dL and is stable. Compute an intravenous digoxin loading and maintenance dose for this patient.
1. Estimate creatinine clearance.
This patient has a stable serum creatinine and is obese [IBWfemales (in kg) = 45 + 2.3 (Ht − 60) = 45 + 2.3 (65 in − 60) = 57 kg]. The Salazar and Corcoran equation can be used to estimate creatinine clearance:

Note: Height is converted from inches to meters: Ht = (65 in • 2.54 cm/in)/(100 cm/m) = 1.65 m.
The drug clearance versus creatinine clearance relationship is used to estimate the digoxin clearance for this patient (ClNR = 20 mL/min since the patient has moderate-severe heart failure):

3. Use average steady-state concentration equation to compute digoxin maintenance dose.
For a patient with heart failure, the desired digoxin concentration would be 0.5-1 ng/mL. A serum concentration equal to 0.8 ng/mL will be chosen for this patient, and intravenous digoxin will be used (F = 1). Note that for concentration units ng/mL = μg/L, and this conversion will be made before the equation is used. Also, conversion factors are needed to change milliliter units to liter (1000 mL/L) and min units to days (1440 min/d).

This intravenous dose would be rounded to 125 μg every other day.
4. Use loading dose equation to compute digoxin loading dose (if needed).
The patient has poor renal function and is obese. Therefore, the volume of distribution equation that adjusts the parameter estimate for renal dysfunction can be used to compute the digoxin loading dose, and ideal body weight will be used as the weight factor. An intravenous loading dose (F = 1) could be given in this patient to achieve the desired pharmacologic effect quicker than would occur if maintenance doses alone were used to allow concentrations to accumulate over 3-5 half-lives.
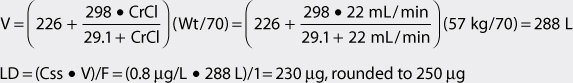
When digoxin loading doses are administered, they are usually given in divided doses separated by 4-6 hours (50% of dose at first, followed by two additional doses of 25%). In this case, an initial intravenous dose of 125 μg would be given initially, followed by two additional intravenous doses of 62.5 μg each. One of the loading doses could be withheld if pulse rate was less than 50-60 beats/min or other undesirable digoxin adverse effects were noted.
Jelliffe Method
Another approach to derive initial doses of digoxin is to compute an appropriate loading dose which provides an amount of the drug in the body that evokes the appropriate pharmacologic response.87,88 The amount of digoxin in the body that produces the desired effect is known at the total body stores (TBS) of digoxin. The percent of drug that is lost on a daily basis (%lost/d) is related to renal function according to the following equation: %lost/d = 14% + 0.20(CrCl), where 14% is the percent of digoxin eliminated per day by nonrenal routes and CrCl is creatinine clearance in mL/min.88 Because the goal of therapy is to provide the total body stores of digoxin that causes the appropriate inotropic or chronotropic effect, the maintenance dose (D in μg/d) is the amount of digoxin eliminated on a daily basis: D = [TBS • (%lost/d)]/F, where TBS is total body stores in μg/d, %lost/d is the percent of digoxin TBS lost per day, F is the bioavailability factor for the dosage form, and 100 is a conversion factor to convert the percentage to a fraction. Combining the two equations produces the initial digoxin maintenance dose: D = {TBS • [14% + 0.20(CrCl)]}/(F•100).
For patients with creatinine clearance values over 30 mL/min, digoxin total body stores of 8-12 μg/kg are usually required to cause inotropic effects while 13-15 μg/kg are generally needed to cause chronotropic effects.89,90 Since renal disease (creatinine clearance < 30 mL/min) decreases digoxin volume of distribution, initial digoxin total body stores of 6-10 μg/kg are recommended for patients with poor renal function.89 Because obesity does not change digoxin volume of distribution, the weight factor used in this calculation is ideal body weight (IBW) for patients that are significantly overweight (> 30% over IBW).33,34 If a patient weighs less than their ideal body weight, actual body weight is used to calculate total body stores. For patients whose weight is between their ideal body weight and 30% over ideal weight, actual body weight can be used to compute total body stores, although some clinicians prefer to use ideal body weight for these individuals. If a loading dose is required, the total body store (TBS in μg) is calculated and used to compute the loading dose (LD in μg) after correction for dosage form bioavailability (F): LD = TBS/F.87,88
Nomograms that use the dosing concepts in the Jelliffe dosing method are available. But, in order to make calculations easier, they make simplifying assumptions. The nomograms are for adults only, and separate versions are needed for intravenous injection (see Table 6-4A) and tablets (see Table 6-4B) because of bioavailability differences among dosage forms. Both nomograms assume that digoxin total body stores of 10 μg/kg are adequate, so are limited to heart failure patients requiring this dose.
To contrast the Jelliffe dosage method with the Jusko-Koup dosage method, the same patient cases will be used as examples for this section.
EXAMPLE 1 
MJ is a 50-year-old, 70-kg (height = 5 ft 10 in) male with atrial fibrillation for less than 24 hours. His current serum creatinine is 0.9 mg/dL, and it has been stable over the last 5 days since admission. Compute an intravenous digoxin dose for this patient to control ventricular rate.
1. Estimate creatinine clearance.
This patient has a stable serum creatinine and is not obese. The Cockcroft-Gault equation can be used to estimate creatinine clearance:
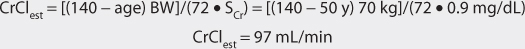
2. Estimate total body store (TBS) and maintenance dose (D).
The patient has good renal function and is nonobese. Digoxin total body stores of 13-15 μg/kg are effective in the treatment of atrial fibrillation. A digoxin dose of 14 μg/kg is chosen for this patient.
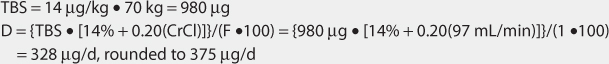
3. Use loading dose equation to compute digoxin loading dose (if needed).
Digoxin total body store is used to calculate the loading dose after correcting for bioavailability:

When digoxin loading doses are administered, they are usually given in divided doses separated by 4-6 hours (50% of dose at first, followed by two additional doses of 25%). In this case, an initial intravenous dose of 500 μg would be given initially, followed by two additional intravenous doses of 250 μg each. One of the loading doses could be withheld if pulse rate was less than 50-60 beats/min or other undesirable digoxin adverse effects were noted.

Same patient profile as in example 1, but serum creatinine is 3.5 mg/dL indicating renal impairment.
1. Estimate creatinine clearance.
This patient has a stable serum creatinine and is not obese. The Cockcroft-Gault equation can be used to estimate creatinine clearance:
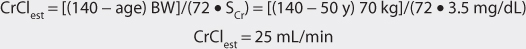
2. Estimate total body store (TBS) and maintenance dose (D).
The patient has poor renal function and is nonobese. Digoxin total body stores of 6-10 μg/kg are recommended for patients with renal dysfunction. A digoxin dose of 8 μg/kg is chosen for this patient.
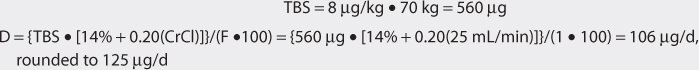
3. Use loading dose equation to compute digoxin loading dose (if needed).
Digoxin total body store is used to calculate the loading dose after correcting for bioavailability:

When digoxin loading doses are administered, they are usually given in divided doses separated by 4-6 hours (50% of dose at first, followed by two additional doses of 25%). In this case, an initial intravenous dose of 250 μg would be given initially, followed by two additional intravenous doses of 125 μg each. One of the loading doses could be withheld if pulse rate was less than 50-60 beats/min or other undesirable digoxin adverse effects were noted.
EXAMPLE 3 
Same patient profile as in example 1, but serum creatinine is 3.5 mg/dL indicating renal impairment. Additionally, the patient is being treated for NYHA Class III moderate heart failure, not atrial fibrillation. Compute an oral digoxin tablet maintenance dose for this patient.
1. Estimate creatinine clearance.
This patient has a stable serum creatinine and is not obese. The Cockcroft-Gault equation can be used to estimate creatinine clearance:
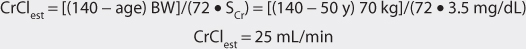
2. Estimate total body store (TBS) and maintenance dose (D).
The patient has poor renal function and is nonobese. Digoxin total body stores of 6-10 μg/kg are recommended for patients with renal dysfunction. A digoxin dose of 8 μg/kg is chosen for this patient.


OI is a 65-year-old, 170-kg (height = 5 ft 5 in) female with NYHA Class III moderate heart failure. Her current serum creatinine is 4.7 mg/dL and is stable. Compute an intravenous digoxin loading and maintenance dose for this patient.
1. Estimate creatinine clearance.
This patient has a stable serum creatinine and is obese [IBWfemales (in kg) = 45 + 2.3 (Ht − 60) = 45 + 2.3 (65 in − 60) = 57 kg]. The Salazar and Corcoran equation can be used to estimate creatinine clearance:

Note: Height is converted from inches to meters: Ht = (65 in • 2.54 cm/in)/(100 cm/m) = 1.65 m.
2. Estimate total body store (TBS) and maintenance dose (D).
The patient has poor renal function and is obese. Digoxin total body stores of 6-10 μg/kg are recommended for patients with renal dysfunction, and ideal body weight (IBW) should be used in the computation. A digoxin dose of 8 μg/kg is chosen for this patient.

This intravenous dose would be rounded to 150 μg every other day.
3. Use loading dose equation to compute digoxin loading dose (if needed).
Digoxin total body store is used to calculate the loading dose after correcting for bioavailability:

When digoxin loading doses are administered, they are usually given in divided doses separated by 4-6 hours (50% of dose at first, followed by two additional doses of 25%). In this case, an initial intravenous dose of 250 μg would be given initially, followed by two additional intravenous doses of 125 μg each. One of the loading doses could be withheld if pulse rate was less than 50-60 beats/min or other undesirable digoxin adverse effects were noted.
USE OF DIGOXIN SERUM CONCENTRATIONS TO ALTER DOSAGES
Because of pharmacokinetic variability among patients, it is likely that doses computed using patient population characteristics will not always produce digoxin serum concentrations that are expected. Because of this, digoxin serum concentrations are measured in many patients to insure that therapeutic, nontoxic levels are present and to check for compliance to dosage regimens. However, not all patients may require serum concentration monitoring. For example, if an appropriate dose for the renal function and concurrent disease states of the patient is prescribed (e.g., 250 μg/d in a patient with a creatinine clearance of 80-100 mL/min for heart failure) and the desired clinical effect is achieved without adverse effects, digoxin serum concentration monitoring may not be necessary. Whether or not digoxin concentrations are measured, important patient parameters (dyspnea, orthopnea, tachypnea, cough, pulmonary rales/edema, S3 gallop, etc) should be followed to confirm that the patient is responding to treatment and not developing adverse drug reactions.
When digoxin serum concentrations are measured in patients and a dosage change is necessary, clinicians should seek to use the simplest, most straightforward method available to determine a dose that will provide safe and effective treatment. In most cases, a simple dosage ratio can be used to change digoxin doses since digoxin follows linear pharmacokinetics. Sometimes, it is not possible to simply change the dose because of the limited number of oral dosage strengths, and the dosage interval must also be changed. Available digoxin tablet strengths are 125 μg and 250 μg while digoxin oral solution is available at a concentration of 0.05 mg/mL. In some situations, it may be necessary to compute the digoxin pharmacokinetic parameters for the patient and utilize these to calculate the best drug dose (Pharmacokinetic Parameter method).
Finally, computerized methods that incorporate expected population pharmacokinetic characteristics (Bayesian pharmacokinetic computer programs) can be used in difficult cases where renal function is changing, serum concentrations are obtained at suboptimal times, or the patient was not at steady-state when serum concentrations were measured. An additional benefit of this dosing method is that a complete pharmacokinetic workup (determination of clearance, volume of distribution, and half-life) can be done with one or more measured concentrations that do not have to be at steady-state.
Linear Pharmacokinetics Method
Because digoxin follows linear, dose-proportional pharmacokinetics, steady-state serum concentrations change in proportion to dose according to the following equation: Dnew/Css,new = Dold/Css,old or Dnew = (Css,new/Css,old)Dold. In this equation, D is the dose in μg, Css is the steady-state concentration in ng/mL, old indicates the dose that produced the steady-state concentration that the patient is currently receiving, and new denotes the dose necessary to produce the desired steady-state concentration. The advantages of this method are that it is quick and simple. The disadvantages are that steady-state concentrations are required. Also, because of a limited number of solid oral dosage strengths, it may not be possible to attain desired serum concentrations by only changing the dose. In these cases, dosage intervals are extended for patients receiving tablets so that doses can be given as multiples of 125 μg. The estimated times to achieve steady-state concentrations on a stable digoxin dosage regimen varies according to renal function and are listed in Tables 6-4A and 6-4B. An alternative to this way of estimating time to steady-state is to compute the expected digoxin half-life (t1/2 in days) for a patient using digoxin clearance (Cl in L/d) and volume of distribution (V in L) and allow 3-5 half-lives to pass before obtaining digoxin serum concentrations: t1/2 = (0.693 • V)/Cl.




