Phase II Drug-Metabolizing Enzymes
LEARNING OBJECTIVES
INTRODUCTION
Role of Phase II Biotransformation in Drug Elimination
There is often a high concordance between the pharmacokinetic properties of drugs and their therapeutic actions. Serum concentrations are frequently used as surrogate markers for the likely concentrations of drugs achieved at the site of action in tissues. Rates of drug biotransformation and elimination are critical factors that influence the serum concentrations of drugs and their duration of action in the body. Rapid drug clearance may lead to subtherapeutic concentrations in tissues that diminishes efficacy. On the other hand, if clearance is impaired, drugs may accumulate and produce toxicity.
Like other xenobiotics, drugs are generally quite hydrophobic and undergo biotransformation to polar products following the concerted actions of several families of drug-metabolizing enzymes; this process enhances clearance. In phase I metabolism a functional group is introduced into the parent molecule that renders the drug more polar. However, the aqueous solubility of many phase I metabolites may be inadequate for efficient elimination. In phase II biotransformation reactions these phase I metabolites of intermediate polarity as well as parent drug undergo conjugation with highly polar endogenous molecules. This markedly increases aqueous solubility and the resultant drug conjugates are eliminated more readily in urine or feces. Thus, phase II biotransformation pathways usually mediate the terminal phase of the removal of drugs and other foreign compounds from the body.
Enzymes of Phase II Biotransformation
There are several classes of phase II biotransformation enzymes that catalyze drug conjugation reactions (Table 6B–1 lists these enzymes and those that mediate the formation of essential cofactors). Among the most important are the UDP-glucuronosyltransferases (UGTs), sulfotransferases (SULTs), N-acetyltransferases (NATs), and glutathione S-transferases (GSTs). Together these enzymes contribute to the majority of clearance pathways that exist for most drugs. Additional phase II enzymes include thiopurine S-methyltransferase (TPMT) and the acyl-CoA synthetase medium-chain family members that act in conjunction with acyl-CoA:amino acid N-acyltransferases to generate amino acid conjugates. While not widely involved in drug biotransformation, some of these enzymes have roles in the clearance of a limited number of therapeutic agents. Finally, there is a group of enzymes such as catechol O-methyltransferase (COMT) and other methyltransferases that are functionally important in the clearance of neurotransmitters or other molecules of endogenous importance. Some studies have suggested that these enzymes participate in drug clearance but the supportive clinical evidence that currently exists is inconclusive.
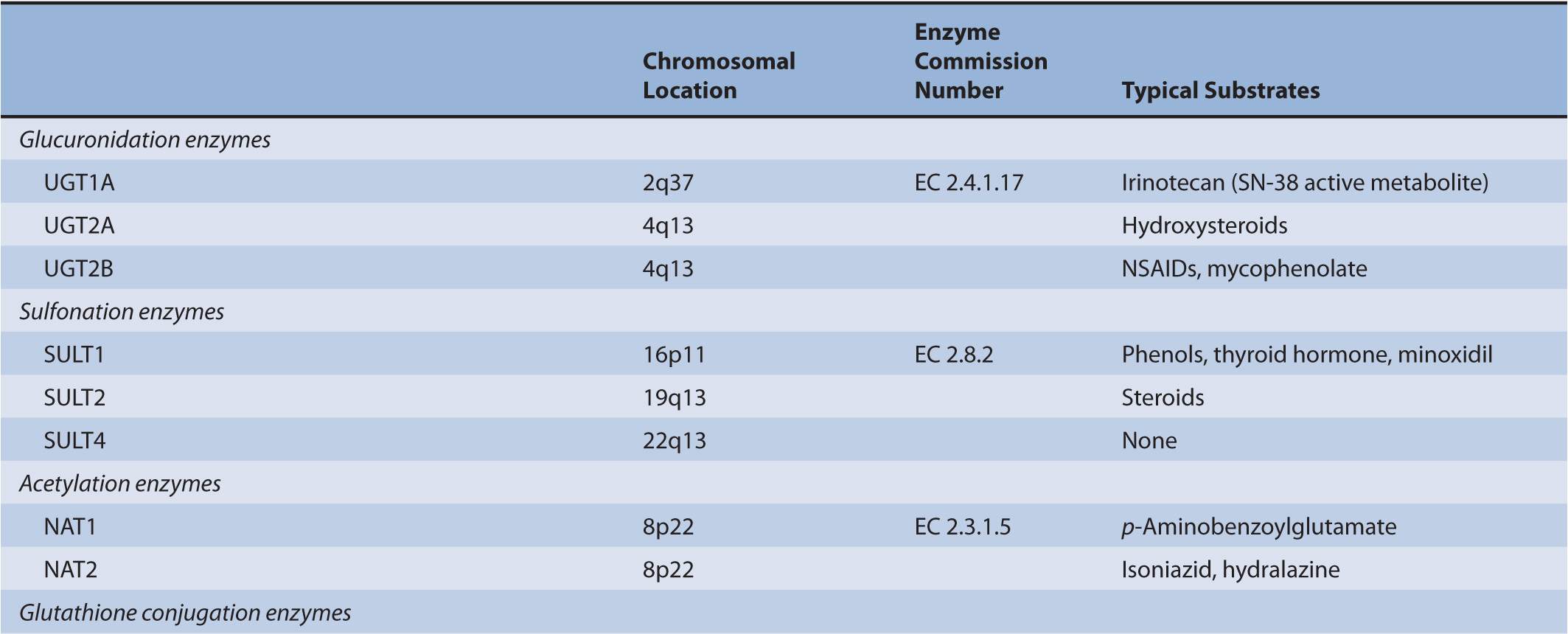
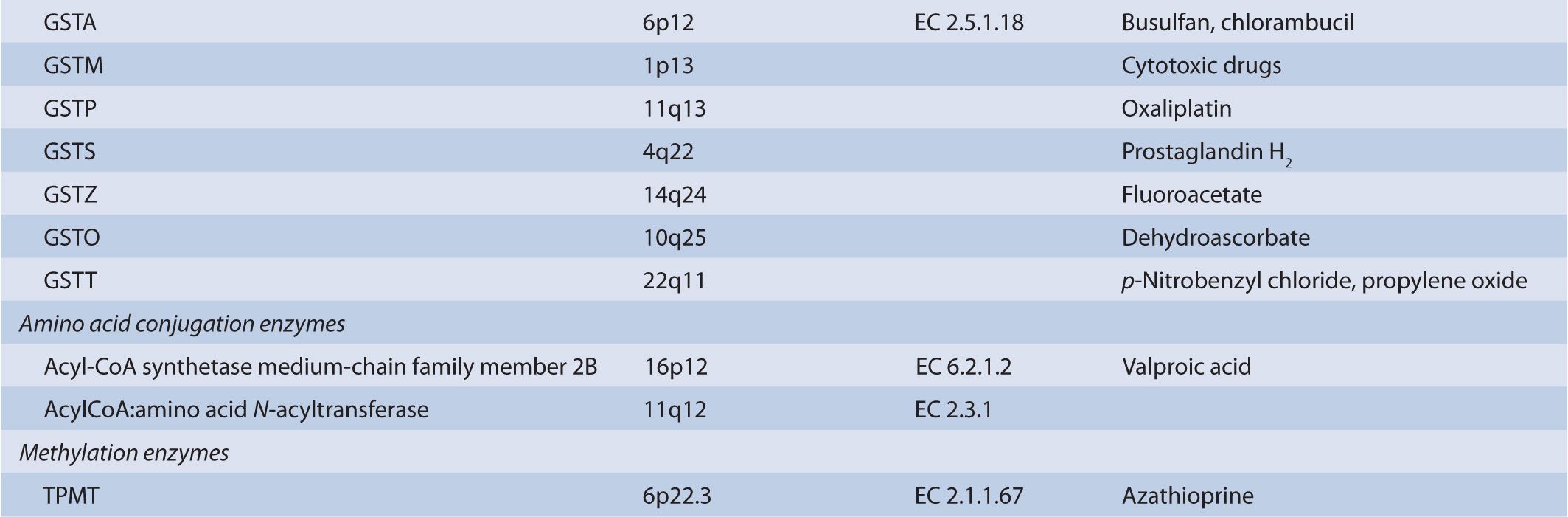
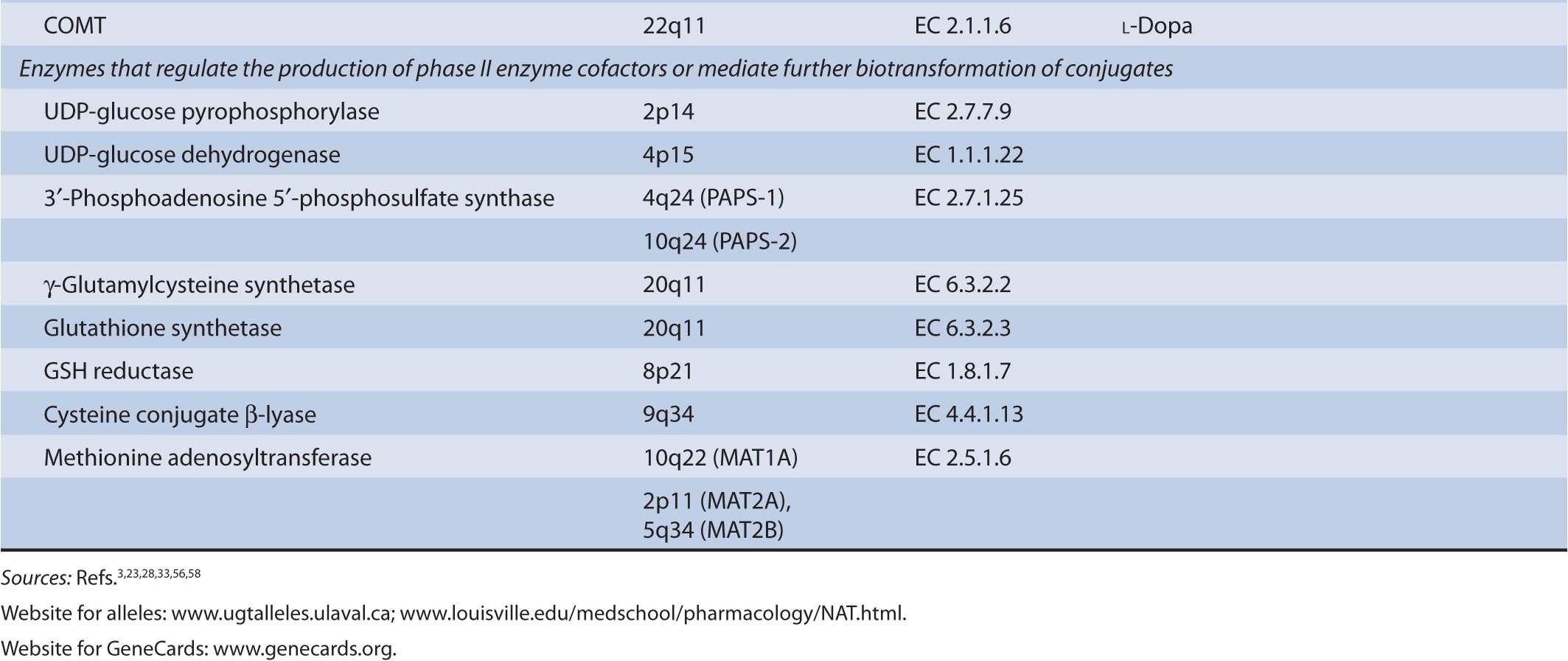
TABLE 6B–1 Genes involved in phase II biotransformation: chromosomal locations, enzyme commission numbers, and typical substrates.
Biotransformation enzymes are frequently involved in pharmacokinetic drug–drug interactions because they accommodate a wide range of substrates that can compete for elimination. In addition, certain allelic variants of genes that encode biotransformation enzymes may be dysfunctional. In both of these situations drug clearance is impaired, which may lead to drug accumulation and may precipitate toxicity. Phase I enzymes typified by the cytochromes P450s (CYPs) usually catalyze the rate-limiting steps in the overall process of drug biotransformation and elimination; CYP-mediated pharmacokinetic drug–drug interactions and defective CYP gene variants are relatively common. By comparison, there are relatively few instances in which pharmacokinetic interactions and phase II pharmacogenetics affect drug clearance. This is due in part to the great reserve capacity for phase II elimination, such that drug interactions and defects in specific phase II genes may be compensated by alternate biotransformation pathways. However, there are several important examples in which the pharmacogenetics of phase II enzymes strongly influences the clearance and toxicity of specific drugs in patients.
Significance of Phase II Gene Polymorphisms
Much of the research to date on the significance of phase II gene polymorphisms has centered around the protective actions of these enzymes against toxic and reactive intermediates generated from carcinogens and aromatic amines. Thus, UGTs, SULTs, NATs, and GSTs are particularly important in the conjugation of reactive intermediates that are generated from such chemicals by the actions of phase I enzymes. Phase II biotransformation usually minimizes the toxicity of reactive species by catalyzing their conversion to stable products that are more hydrophilic and readily eliminated, for example, CYPs activate polyaromatic hydrocarbons such as benzo(a)pyrene to reactive epoxide intermediates that are able to bind tightly to DNA; phase II biotransformation converts these species to conjugates that are less toxic.
Drug therapy is usually initiated using a limited range of dosage regimen, with subsequent dose optimization undertaken empirically, based on observed toxicity and efficacy in individuals (although the latter may be difficult to measure). However, the desired therapeutic effects do not occur uniformly in all patients when drugs are administered according to standard regimen; many subjects are either underdosed or overdosed. Pharmacogenetic approaches, in which patient-specific factors, such as the presence of defective phase II alleles that may impact on drug pharmacokinetics, could be used to direct drug dosage and provide opportunities to tailor therapy to individuals. This chapter will focus on well-defined and emerging examples of the impact of allelic variation in genes that encode phase II drug-metabolizing enzymes that influence the clinical efficacy and safety of drugs. These factors are of greatest impact when they mediate a critical pathway of elimination of a drug or metabolite that has a major toxicity profile and for which the alternate pathways of elimination are limited.
IMPORTANT ENZYMES THAT MEDIATE PHASE II DRUG BIOTRANSFORMATION IN MAN
UGTs
Drug Glucuronidation by UGT Enzymes
UGT enzymes (EC 2.4.1.17) catalyze the glucuronidation of polar aglycones that contain phenolic, alcoholic, or carboxylic oxygen atoms and a limited number of nitrogen- and sulfur-containing molecules. The enzymes are active on a diverse range of drugs and xenobiotic substrates, as well as endobiotics such as bilirubin, steroid, and thyroid hormones. UGT activity is supported by the cofactor UDP-glucuronic acid (uridine-5′-diphospho-α-D-glucuronic acid) that is synthesized from glucose-1-phosphate and uridine triphosphate via the sequential actions of UDP-glucose pyrophosphorylase (EC 2.7.7.9) and UDP-glucose dehydrogenase (EC 1.1.1.22). Nucleophilic attack of the aglycone substrate on UDP-glucuronic acid occurs with inversion of configuration and produces the corresponding β-glucuronides. Kinetic studies have suggested that UGTs operate via an SN2-like mechanism involving base catalysis.1 Thus, within the active center of the UGT enzyme His and Asp residues facilitate the deprotonation or polarization of a suitable substituent in an aglycone substrate, which facilitates nucleophilic attack at C-1 of glucuronic acid. Lone electron pairs on tertiary amine substrates are sufficiently nucleophilic to generate glucuronides without the requirement for bond polarization.
UGTs are integral proteins of the endoplasmic reticulum and membrane lipid composition is a critical determinant of rates of xenobiotic glucuronidation. UGTs are oriented toward the lumen of the endoplasmic reticulum with a short C-terminal tail facing the cytoplasmic side. It has been proposed that dimerization of different UGT monomers may lead to the creation of a quasi–substrate recognition site that augments the metabolic capacity and substrate specificity of UGTs beyond that seen with the monomers.2
Multiplicity of the Human UGT System
Major human drug-metabolizing UGTs are encoded by three gene subfamilies: UGT1A, UGT2A, and UGT2B. The UGT2B subfamily, which comprises several genes, and the UGT2A1 gene are located on chromosome 4. In contrast, the entire UGT1 family is derived from a single locus of 210 kilobase (kb) on chromosome 2 (2q37). UGT1A genes are composed of 5 exons, with 1 of 13 alternate exons comprising exon 1 and exons 2–5 being common to all UGT1As.
A number of human UGTs have been identified to date: UGT1A1, 1A3–1A10, 2A1, 2A2 (which may be an isoform of UGT2A1), 2B4, 2B7, 2B10, 2B11, 2B15, 2B17, and 2B28.3 All but UGT1A7, 1A8, and 1A10 are expressed in liver, while those three enzymes are expressed exclusively in the gastrointestinal tract, and UGT2A1 is expressed in the nasal epithelium where it is involved in the termination of signaling by odorant molecules.3 UGTs 1A1, 1A3, 1A4, 1A6, 1A9, 2B7, and 2B15 contribute to hepatic drug elimination, but UGT2B7 is functionally the most important enzyme in drug glucuronidation because it accommodates a diverse range of substrates, including opioids, nonsteroidal anti-inflammatory drugs (NSAIDs), analgesics, antiepileptic agents, oncology agents, antiretrovirals, and hypolipidemic agents. UGTs 1A7, 1A8, and 1A10 may contribute to local drug biotransformation in the gastrointestinal tract.
To date many polymorphisms have been described for both UGT1 and UGT2B genes, but how UGT pharmacogenetics affects biotransformation and clearance has been defined for only a limited number of drugs (Table 6B–2). Compared with phase I CYPs there are few UGT-selective substrates that represent major toxicity concerns. Despite this general point, there remain several drugs for which defective UGT alleles are important determinants of clinical toxicity.
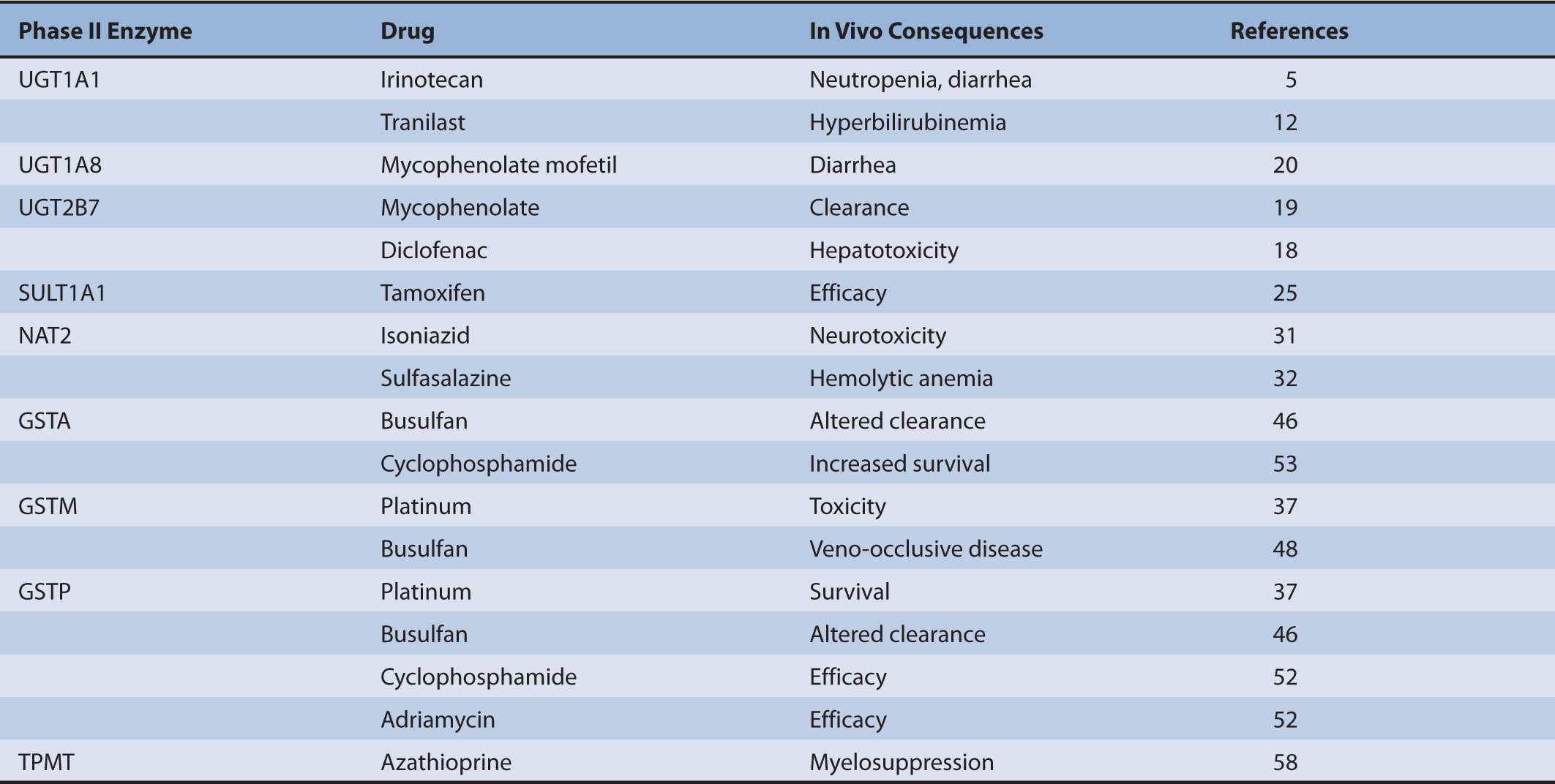
TABLE 6B–2 Pharmacogenetics of phase II biotransformation enzymes: impact on drug clearance and therapeutic outcomes.
Role of UGT1A1 in Clearance of Bilirubin and Irinotecan
UGT1A1 has a major role in the glucuronidation of the heme degradation product bilirubin. Defects in the UGT1A1 gene impair bilirubin conjugation, leading to hyperbilirubinemias of varying severity.4 Three levels of UGT1A1 deficiency have been identified. Crigler–Najjar syndrome type 1 is most severe and is characterized by potentially lethal hyperbilirubinemia (serum bilirubin >0.34 mM) that occurs in the absence of hemolysis or other liver disease. Crigler–Najjar syndrome type 2 is an intermediate form of hyperbilirubinemia (serum bilirubin ~-0.12–0.34 mM) characterized by residual UGT1A1 activity and Gilbert syndrome is the mildest form in which serum bilirubin levels fluctuate up to 0.085 mM. In Caucasian, African American, and South Asian populations the exon sequences of the UGT1A1 gene are normal but the TATAA element in the 5′-upstream region exhibits a variable number of tandem repeat polymorphism. Thus, the UGT1A1*28 promoter variant carries the sequence A(TA)7TAA in place of the wild-type A(TA)6TAA (Figure 6B–1), which decreases the transcription rate of the UGT1A1 gene and results in defective bilirubin glucuronidation. Around 10% of Caucasians are homozygous and about 40% are heterozygous for the UGT1A1*28 variant. In contrast, a homozygous nonsynonymous polymorphism that encodes the Gly71Arg amino acid replacement (the *6 variant) is the common cause of the Gilbert phenotype in East Asian subjects (allele frequency ~20%).5 Although these common UGT1A1 variants occur with differing frequency in different ethnic groups, additional UGT1A1 variants have also been detected by resequencing the UGT1A1 gene in subjects with Crigler–Najjar syndrome.
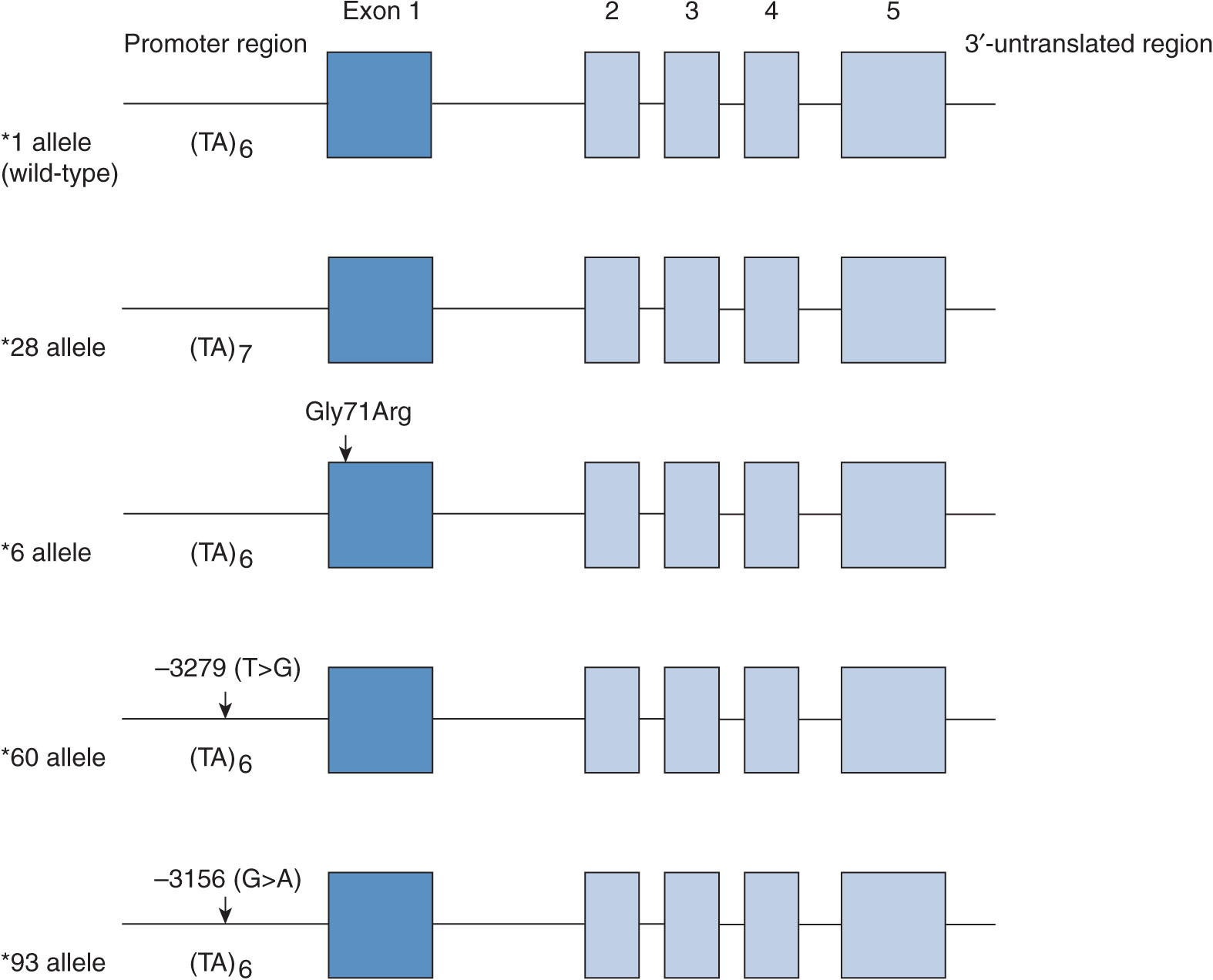
FIGURE 6B–1 Gene structure of UGT1A1 and major variants associated with impaired glucuronidation of SN-38, the pharmacologically active metabolite of irinotecan. The gene contains five exons, an important promoter sequence in the 5′-untranslated region, and intronic and 3′-untranslated regions. The *28, *60, and *93 alleles carry polymorphisms in the TATAA box or adjacent regulatory sequences, whereas the *6 allele contains a polymorphism in exon 1 that leads to the Gly71Arg amino acid substitution. (The gene structure is not drawn to scale.)
The conjugation and elimination of drugs may also be affected in individuals who carry UGT1A1 polymorphisms (Table 6B–1). Irinotecan (CPT-11 or (S)-4,11-diethyl-3,4,12,14-tetrahydro-4-hydroxy-3,14-dioxo-1H-pyrano(3′,4′:6,7)-indolizino(1,2-b) quinolin-9-yl-(1,4′-bipiperidine)-1′-carboxylate) is a topoisomerase II inhibitor that is the first-line treatment for metastatic colorectal cancer. It is a prodrug that is converted by carboxylesterases to the active metabolite SN-38 (7-ethyl-10-hydroxycamptothecin; Figure 6B–2).5 SN-38 undergoes UGT1A1-mediated glucuronidation to 10-O-glucuronyl-SN-38 (SN-38G), which limits the duration of action of the drug. Thus, UGT1A1 activity is a major factor that influences the extent of SN-38G formation and irinotecan toxicity. Indeed, the plasma ratio of SN-38G:SN-38 has been related to the incidence of neutropenia and diarrhea. In individuals with defective glucuronidation SN-38 accumulates and elicits neutropenia and diarrhea. Severe grade diarrhea induced by irinotecan does not respond to loperamide treatment and may necessitate hospitalization, as well as irinotecan dose interruption. Mortality from irinotecan therapy is significant in up to 5% of patients who receive single-agent therapy, but some improvements have been obtained by including 5-fluorouracil and leucovorin in a combination approach.
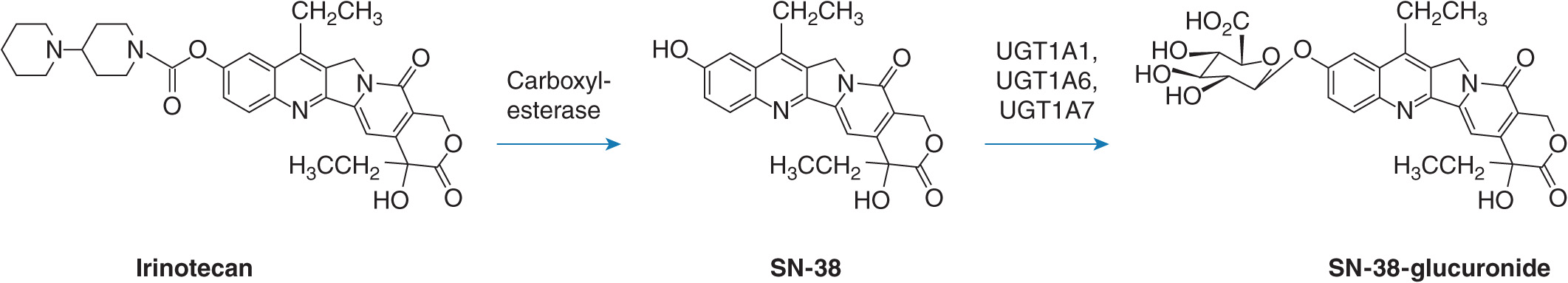
FIGURE 6B–2 Biotransformation of irinotecan to SN-38 and SN-38 glucuronide mediated by carboxylesterases and UGTs, respectively.
A large number of clinical studies have shown that the UGT1A1*28 allele confers susceptibility to irinotecan toxicity, which has led the US FDA to recommend a dose reduction for irinotecan in individuals who are homozygous for this variant (http://www.fda.gov/medwatch/SAFETY/2005/Jun_PI/Camptosar_PI.pdf).6 This would decrease toxicity in patients who have a major glucuronidation defect, but specific guidelines on the actual dose reduction that should be applied or whether heterozygotes should also receive a dose reduction are currently lacking.5 Other UGT1A1 allelic variants that impair SN-38 glucuronidation are more prevalent in other populations and may also necessitate irinotecan dose adjustments. In a dose-escalation study Satoh et al.7 administered irinotecan (75, 100, or 150 mg/m2) to individuals of differing genotype: homozygous wild-type (*1/*1), heterozygous variants (*1/*6 or *1/*28), or homozygous variants (*6/*6, *28/*28, or *6/*28). In patients who progressed to the highest dose, the incidence of dose-limiting toxicities was greatest in those carrying the homozygous variant genotype. Because there is a similar incidence of the UGT1A1*6 and *28 variants in East Asians (allele frequencies 10–25%) testing for both alleles has been approved in Japan for use in current oncology practice.5
The association between UGT1A1 gene defects and impaired conjugation of both bilirubin and SN-38 suggests that hyperbilirubinemia might be a predictor of irinotecan toxicity. However, there is increasing evidence that the relationship between UGT1A1 gene function and irinotecan toxicity may be more complex. Indeed, close associations between the UGT1A1*28 polymorphism and predisposition to irinotecan toxicity have been observed in some but not all studies. In part this may be due to the apparent capacity of other UGTs to detoxify SN-38. Thus, the affinities of the hepatic UGT1A6 and extrahepatic UGT1A7 for SN-38 are several-fold greater than that of UGT1A1.8,9 These UGTs may also contribute to SN-38 glucuronidation and pharmacogenetic variation in UGT1A6/1A7 may influence irinotecan safety in patients.
The first haplotype analysis of a regulatory module of ~600 base pair (bp) in the UGT1A1 5′-flanking region that contained the UGT1A1*28 polymorphism was conducted by Innocenti et al.10 using samples from 55 Caucasians and 37 African Americans. This region was selected because it is implicated in basal regulation of the UGT1A1 gene and because it was known to be highly polymorphic. Close linkages between several polymorphisms were identified, including –3279T > G (*60) and -3156G > A (*93) as well as A(TA)7TA (*28), especially in Caucasians; the frequencies of inferred haplotypes varied somewhat between ethnic groups.10,11 However, of the most common haplotypes in Caucasians and African Americans, the only major difference was in the incidence of the *60 polymorphism. Subsequently, another haplotype analysis in 195 Japanese subjects sequenced all 5 exons, the promoter and enhancer regions, and all flanking introns. Four haplotype groups based on the *1, *60, *6, and *28 alleles were identified that had similar frequencies.11 Interestingly, all of these haplotypes were associated with a significant decrease in the serum SN-38G/SN-38 concentration ratio, which provides evidence that multiple combinations of polymorphisms in the UGT1A1 gene are associated with decreased capacity to inactivate SN-38 by glucuronidation.
UGT Pharmacogenetics and the Incidence of Drug-Induced Hyperbilirubinemia
Several studies suggest that individuals with Gilbert syndrome who carry UGT1A1 polymorphisms may be susceptible to drug-induced hyperbilirubinemias. Thus, tranilast-mediated hyperbilirubinemia was more prevalent in carriers of UGT1A1*2812 and plasma concentrations of the HIV-protease inhibitor atazanavir correlated directly with plasma bilirubin concentrations.13 Indeed, there was a higher incidence of jaundice in patients who received atazanavir or indinavir and who were homozygous for UGT1A1*28.14 Although atazanavir does not undergo extensive glucuronidation, it has been found to inhibit UGTs 1A1, 1A3, and 1A4.15 This may account for clinical findings in which atazanavir-mediated inhibition of hepatic UGT activity increased serum bilirubin levels in most subjects. This was especially pronounced in the one third of individuals who carried variant UGT1A alleles that included UGT1A1*28, UGT1A7*12 and UGT1A7*2, and the UGT1A3-66C promoter polymorphism that is found in several variant alleles of the UGT1A3 gene.16 Indeed, homozygous combinations of UGT1A1*28 and one of the other single nucleotide polymorphisms significantly increased the susceptibility to druginduced hyperbilirubinemia. From this study it was suggested that a haplotype spanning three UGT1A genes could predispose to atazanavir-mediated hyperbilirubinemia, and may therefore constitute a pharmacogenomic risk factor for therapy with the drug.16 Taken together, it is emerging that potent UGT1A inhibitors may impair bilirubin glucuronidation that leads to hyperbilirubinemia. The clinical consequences seem to be most pronounced in carriers of defective UGT1A1 alleles in whom glucuronidation capacity is already compromised. It will now be of interest to evaluate whether other HIV-protease inhibitors carry a similar risk of drug-induced hyperbilirubinemia in patients. This would provide insight into whether genotyping for UGT1A1*28 or other UGT1A gene variants before initiation of antiretroviral therapy might assist in identifying HIV-infected individuals who are at risk from drug-induced jaundice.
Clinical Significance of UGT2B7 Polymorphisms
UGT2B7 is an important catalyst in the glucuronidation of a wide range of drug substrates, including NSAIDs (Table 6B–2). The common C802T polymorphism that encodes the His268Tyr amino acid substitution (the UGT2B7*2 allele) is present in around one third of the Caucasian population; the incidence is lower in Asians.17 The NSAID diclofenac undergoes extensive glucuronidation mediated by UGT2B7 and elicits idiosyncratic hepatotoxicity that causes significant mortality if it progresses to jaundice. Low-activity variants of UGT2B7 appear to predispose to an increased risk of hepatotoxicity from reactive diclofenac metabolites.18 This finding suggests that UGT2B7 plays an important protective role by mediating the conjugation and clearance of diclofenac that might otherwise undergo biotransformation to hepatotoxic reactive metabolites.
Mycophenolate mofetil is an immunosuppressive drug that prevents the rejection of transplanted organs and stem cells. The drug is an ester that undergoes esterase-mediated hydrolysis to the active metabolite mycophenolate, which is a substrate for several UGTs. In recent studies pediatric renal transplant patients who carried the UGT2B7*2 variant (either homozygotes or heterozygotes) were found to clear mycophenolate less efficiently than wild-type subjects.19 In that study a population pharmacokinetic model was developed that described mycophenolate dosage in individuals in terms of patient body weight, concomitant medication, and UGT2B7 genotype.
UGT genotype may be an important determinant of gastrointestinal side effects due to mycophenolate mofetil. Diarrhea is a significant adverse effect of the drug that is reportedly less likely in patients who carry the UGT1A8*2 variant allele.20 The mechanism underlying this adverse effect is currently unclear but some studies suggest that it may be due to the acyl glucuronide of the drug. Because UGT1A8 is expressed in the gastrointestinal tract, it is feasible that a decrease in the local formation of the acyl glucuronide in carriers of the low-activity allele could mediate improved tolerance to the drug. However, there are several other considerations that may be pertinent. First, mycophenolate also generates a phenol glucuronide that is inactive so that the relative formation of the acyl and phenolic glucuronides may influence the incidence of toxicity. Second, other UGTs such as UGT2B7 contribute to mycophenolate clearance and, third, ATP-binding cassette efflux transporters are important determinants of mycophenolate elimination in bile, which would deliver mycophenolate conjugates to the gastrointestinal tract. It is likely that the interplay of these factors may ultimately be found to mediate the toxicity and elimination of mycophenolate.
SULTs
Sulfonylation of Drugs by SULT Enzymes
Cytosolic SULTs (EC 2.8.2) catalyze the transfer of the sulfonyl group of 3′-phosphoadenosine 5′-phosphosulfate (PAPS) to hydroxyl, sulfhydryl, amino, or N-oxide substituents in suitable endobiotics and xenobiotics and generates sulfate conjugates that are readily excreted in urine. The cofactor PAPS is itself synthesized from ATP and inorganic sulfate by the bifunctional 3′-phosphoadenosine 5′-phosphosulfate synthase (PAPSS; EC 2.7.1.25) that exists as two isoforms—PAPSS-1 and PAPSS-2—that are abundant in brain and liver, respectively. Sulfation is a lower-capacity pathway than glucuronidation. Thus, many drugs and other xenobiotics are substrates for both SULTs and UGTs, but UGT-dependent pathways usually predominate.
There are two broad classes of SULTs: the membrane-bound SULTs that are located in the Golgi apparatus and that mediate the sulfonation of proteins, lipids, and glycosaminoglycans, and the cytosolic SULTs that mediate the sulfonation of drugs, other xenobiotics, and endogenous substrates that include steroids, bile acids, and neurotransmitters. Kinetic studies have suggested that sulfonation follows an ordered Bi Bi mechanism via an SN2 nucleophilic substitution reaction in which a lone pair of electrons on the substrate oxygen attacks the activated sulfur atom in PAPS with displacement of the phosphate group.21 Most of the cytosolic SULTs appear to exist as dimers; these may be either homodimers or heterodimers.
Multiplicity of the Human SULT System
To date, three gene families have been identified in humans: SULT1 (located on chromosome 16), SULT2 (chromosome 19), and SULT4 (chromosome 22). These families contain multiple genes that encode 13 distinct enzymes: SULT1—A1, A2, A3, A4, B1, C2, C4, and E1; SULT2—A1 and B1, and SULT4A1.22 Both SULT2B1 and SULT4A1 encode a pair of isoforms—SULT2B1-v1 and SULT2B1-v2—that differ only in their N-terminal sequences as a result of either alternate promoter usage or alternate mRNA splicing,22 while the SULT4A1-v1 and SULT4A1-v2 isoforms differ in their C-terminal amino acid sequences. The human SULTs that mediate the sulfonylation of endobiotics and xenobiotics containing phenolic and alcoholic hydroxyl groups belong to families 1 and 2, while the function of human SULT4A1 is currently unknown.
Quantitatively, SULT1A1 is the major SULT1A expressed in adult liver and has also been detected in numerous extrahepatic tissues. By contrast, SULT1A3 is highly expressed in extrahepatic tissues but only at low level in liver. The tissue distribution and physiological function of SULT1A2 is not well understood because although the mRNA has been detected in numerous tissues, the protein has not.22
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


