Pharmacotherapy of the Epilepsies
Epileptic seizures often cause transient impairment of consciousness, leaving the individual at risk of bodily harm and often interfering with education and employment. Therapy is symptomatic: Available drugs inhibit seizures, but neither effective prophylaxis nor cure is available. The mechanisms of action of antiseizure drugs fall into 3 major categories:
1. Limiting the sustained, repetitive firing of neurons, an effect mediated by promoting the inactivated state of voltage-activated Na+ channels
2. Enhancing synaptic inhibition mediated γ-aminobutyric acid (GABA), a drug effect that may occur via presynaptic or postsynaptic action
3. Inhibition of voltage-activated Ca2+ channels responsible for T-type Ca2+ currents
Drugs effective against the most common forms of epileptic seizures, partial and secondarily generalized tonic-clonic seizures, appear to work by one of the first two mechanisms. Drugs effective against the less common absence seizure work by the third mechanism.
TERMINOLOGY AND EPILEPTIC SEIZURE CLASSIFICATION
The term seizure refers to a transient alteration of behavior due to the disordered, synchronous, and rhythmic firing of populations of brain neurons. The term epilepsy refers to a disorder of brain function characterized by the periodic and unpredictable occurrence of seizures. Seizures are thought to arise from the cerebral cortex, not from other CNS structures. Epileptic seizures are classified as partial seizures, those beginning focally in a cortical site, and generalized seizures, those that involve both hemispheres widely from the outset. The behavioral manifestations of a seizure are determined by the functions normally served by the cortical site at which the seizure arises. For example, a seizure involving motor cortex is associated with clonic jerking of the body part controlled by this region of cortex. A simple partial seizure is associated with preservation of consciousness. A complex partial seizure is associated with impairment of consciousness. The majority of complex partial seizures originate from the temporal lobe. Examples of generalized seizures include absence, myoclonic, and tonic-clonic. The type of epileptic seizure is one determinant of the drug selected for therapy. Table 21–1 presents more detailed information on the classification of seizures and available medications.
Table 21–1
Classification of Epileptic Seizures

More than 50 distinct epileptic syndromes have been identified and categorized into partial versus generalized epilepsies. The partial epilepsies account for roughly 60% of all epilepsies. The etiology commonly consists of a lesion in some part of the cortex, such as a tumor, developmental malformation, or damage due to trauma or stroke. The generalized epilepsies account for ~40% of all epilepsies and the etiology is usually genetic. The most common generalized epilepsy, referred to as juvenile myoclonic epilepsy, accounts for ~10% of all epileptic syndromes. Like most of the generalized-onset epilepsies, juvenile myoclonic epilepsy is a complex genetic disorder that is probably due to inheritance of multiple susceptibility genes.
NATURE AND MECHANISMS OF SEIZURES AND ANTISEIZURE DRUGS
PARTIAL EPILEPSIES
Either reduction of inhibitory synaptic activity or enhancement of excitatory synaptic activity might be expected to trigger a seizure; pharmacological studies of seizures support this notion. The neurotransmitters mediating the bulk of synaptic transmission in the mammalian brain are amino acids, with GABA and glutamate being the principal inhibitory and excitatory neurotransmitters, respectively (see Chapter 14). Antagonists of the GABAA receptor or agonists of different glutamate-receptor subtypes (NMDA, AMPA, or kainic acid) trigger seizures in experimental animals in vivo. Conversely, drugs that enhance GABA-mediated synaptic inhibition or glutamate-receptor antagonists inhibit seizures in diverse models.
Electrophysiological analyses of individual neurons during a partial seizure demonstrate that the neurons undergo depolarization and fire action potentials at high frequencies (Figure 21–1). This pattern of neuronal firing is characteristic of a seizure and is uncommon during physiological neuronal activity. Thus, selective inhibition of this pattern of firing would be expected to reduce seizures with minimal unwanted effects. Inhibition of the high-frequency firing may be mediated by reducing the ability of Na+ channels to recover from inactivation (Figure 21–2). Depolarization-triggered opening of the Na+ channels in the axonal membrane of a neuron is required for an action potential; after opening, the channels spontaneously close, a process termed inactivation. This inactivation is thought to cause the refractory period during which it is not possible to evoke another action potential. Because firing at a slow rate permits sufficient time for Na+ channels to recover from inactivation, inactivation has little or no effect on low-frequency firing. However, reducing the rate of recovery of Na+ channels from inactivation would limit the ability of a neuron to fire at high frequencies, an effect that likely underlies the effects of carbamazepine, lamotrigine, phenytoin, topiramate, valproic acid, and zonisamide against partial seizures.
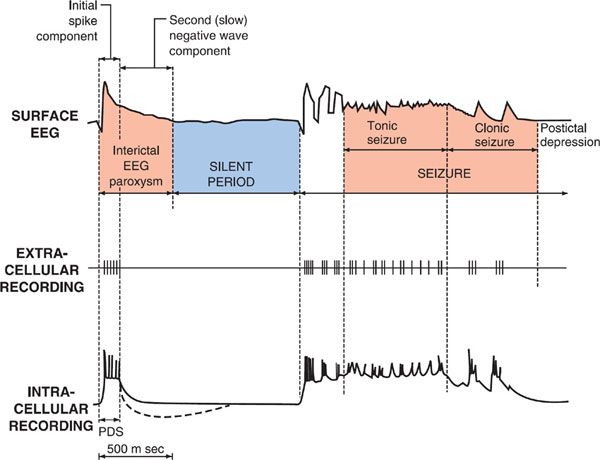
Figure 21–1 Cortical EEG, extracellular, and intracellular recordings in a seizure focus induced by local application of a convulsant agent to mammalian cortex. The extracellular recording was made through a high-pass filter. Note the high-frequency firing of the neuron evident in both extracellular and intracellular recording during the paroxysmal depolarization shift (PDS). (Modified with permission from Ayala GF, Dichter M, Gumnit RJ, et al. Genesis of epileptic interictal spikes. New knowledge of cortical feedback systems suggests a neurophysiological explanation of brief paroxysms. Brain Res, 1973;52:1–17. Copyright © Elsevier.)
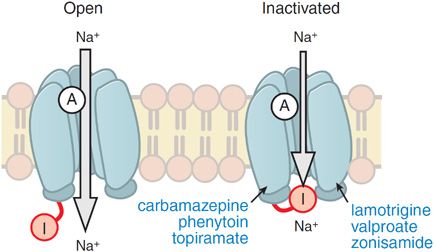
Figure 21–2 Antiseizure drug-enhanced Na+ channel inactivation. Some antiseizure drugs (shown in blue text) prolong the inactivation of the Na+ channels, thereby reducing the ability of neurons to fire at high frequencies. Note that the inactivated channel itself appears to remain open, but is blocked by the inactivation gate I. A, activation gate.
Enhancing GABA-mediated synaptic inhibition would reduce neuronal excitability and raise the seizure threshold. Several drugs are thought to inhibit seizures by regulating GABA-mediated synaptic inhibition through an action at distinct sites of the synapse. The principal postsynaptic receptor of synaptically released GABA is termed the GABAA receptor (see Chapters 14 and 17). Activation of the GABAA receptor inhibits the postsynaptic cell by increasing the inflow of Cl– ions into the cell, which tends to hyperpolarize the neuron. Clinically relevant concentrations of both benzodiazepines and barbiturates enhance GABAA receptor–mediated inhibition through distinct actions on the GABAA receptor (Figure 21–3), and this enhanced inhibition probably underlies the effectiveness of these compounds against partial and tonic-clonic seizures in humans. At higher concentrations, such as might be used for status epilepticus, these drugs also can inhibit high-frequency firing of action potentials. A second mechanism of enhancing GABA-mediated synaptic inhibition is thought to underlie the antiseizure mechanism of tiagabine; tiagabine inhibits the GABA transporter, GAT-1, and reduces neuronal and glial uptake of GABA.
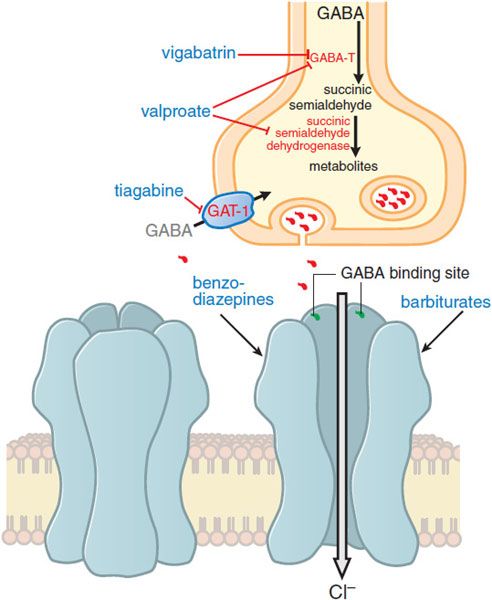
Figure 21–3 Enhanced GABA synaptic transmission. In the presence of GABA, the GABAA receptor (structure on left) is opened, allowing an influx of Cl–, which in turn increases membrane polarization. Some antiseizure drugs (show in larger blue text) act by reducing the metabolism of GABA. Others act at the GABAA receptor, enhancing Cl– influx in response to GABA. As outlined in the text, gabapentin acts presynaptically to promote GABA release; its molecular target is currently under investigation. GABA molecules; GABA-T, GABA transaminase; GAT-1, GABA transporter.
GENERALIZED-ONSET EPILEPSIES: ABSENCE SEIZURES
In contrast to partial seizures, which arise from localized regions of the cerebral cortex, generalized-onset seizures arise from the reciprocal firing of the thalamus and cerebral cortex. Among the diverse forms of generalized seizures, absence seizures have been studied most intensively. The EEG hallmark of an absence seizure is generalized spike-and-wave discharges at a frequency of 3 per second (3 Hz). EEG spikes are associated with the firing of action potentials and the following slow wave with prolonged inhibition. These reverberatory, low-frequency rhythms are made possible by a combination of factors, including reciprocal excitatory synaptic connections between the neocortex and thalamus. One intrinsic property of thalamic neurons that is pivotally involved in the generation of the 3-Hz spike-and-wave discharges is the low threshold (“T-type”) Ca2+ current. T-type currents amplify thalamic membrane potential oscillations, with one oscillation being the 3-Hz spike-and-wave discharge of the absence seizure. Importantly, the principal mechanism by which anti–absence-seizure drugs (ethosuximide, valproic acid) are thought to act is by inhibition of the T-type Ca2+ channels (Figure 21–4). Thus, inhibiting voltage-gated ion channels is a common mechanism of action among antiseizure drugs, with anti–partial-seizure drugs inhibiting voltage-activated Na+ channels and anti–absence-seizure drugs inhibiting voltage-activated Ca2+ channels.
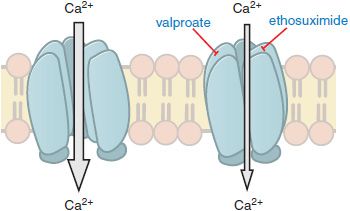
Figure 21–4 Antiseizure drug-induced reduction of current through T-type Ca2+ channels. Some antiseizure drugs (shown in blue text) reduce the flow of Ca2+ through T-type Ca2+ channels, thereby reducing the pacemaker current that underlies the thalamic rhythm in spikes and waves seen in generalized absence seizures.
GENETIC APPROACHES TO THE EPILEPSIES. Genetic causes are solely responsible for rare epileptic forms inherited in an autosomal dominant or autosomal recessive manner. Genetic causes also are mainly responsible for more common forms such as juvenile myoclonic epilepsy (JME) or childhood absence epilepsy (CAE), the majority of which are likely due to inheritance of 2 or more susceptibility genes. Genetic determinants also may contribute some degree of risk to epilepsies caused by injury of the cerebral cortex. Because most patients with epilepsy are neurologically normal, elucidating the mutant genes underlying familial epilepsy in otherwise normal individuals is of particular interest. This has led to the identification of 25 distinct genes implicated in distinct idiopathic epilepsy syndromes that account for <1% of all of the human epilepsies. Almost all of the mutant genes encode voltage- or ligand-gated ion channels. Mutations have been identified in Na+, K+, Ca2+, and Cl– channels, in channels gated by GABA and acetylcholine, and most recently, in intracellular Ca2+ release channels (RyR2) activated by Ca2+. The genotype-phenotype correlations of these genetic syndromes are complex. The cellular electrophysiological consequences of these mutations help describe the mechanisms of seizures and antiseizure drugs. For example, generalized epilepsy with febrile seizures (GEFS+) is caused by a point mutation in the β subunit of a voltage-gated Na+ channel (SCN1B). The phenotype of the mutated Na+ channel appears to involve defective inactivation.
ANTISEIZURE DRUGS: GENERAL CONSIDERATIONS
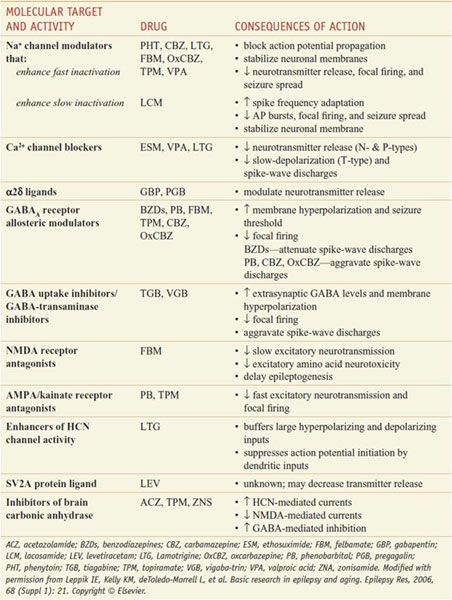
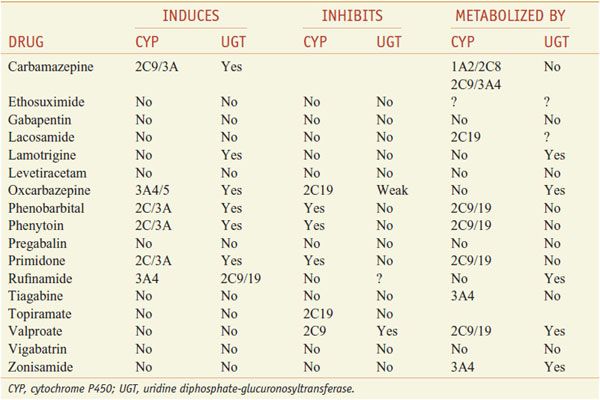
Table 21–2 presents proposed mechanisms of action of antiseizure drugs, classified according to likely molecular target and activity. It is important to select the appropriate drug or combination of drugs that best controls seizures in an individual patient at an acceptable level of untoward effects. As a general rule, complete control of seizures can be achieved in up to 50% of patients, while another 25% can be improved significantly. The degree of success varies as a function of seizure type, cause, and other factors. Drugs used currently frequently cause unwanted effects that range in severity from minimal impairment of the CNS to ideation and attempt of suicide to death from aplastic anemia or hepatic failure. To minimize toxicity, treatment with a single drug is preferred. If seizures are not controlled with the initial agent at adequate plasma concentrations, substitution of a second drug is preferred to the concurrent administration of another agent. However, multiple-drug therapy may be required, especially when 2 or more types of seizure occur in the same patient. Measurement of drug concentrations in plasma facilitates optimizing antiseizure medication. However, clinical effects of some drugs do not correlate well with their concentrations in plasma. The ultimate therapeutic regimen must be determined by clinical assessment of effect and toxicity. Many of these agents interact noticeably with other medications via induction or inhibition of hepatic CYPs and UGTs that are responsible for drug metabolism (Table 21–3).
Table 21–2
Proposed Mechanisms of Action of Antiseizure Drugs
Interactions of Antiseizure Drugs with Hepatic Microsomal Enzymes
HYDANTOINS
PHENYTOIN
Phenytoin (DILANTIN) is effective against all types of partial and tonic-clonic seizures but not absence seizures.
PHARMACOLOGICAL EFFECTS. Phenytoin exerts antiseizure activity without causing general depression of the CNS. In toxic doses, it may produce excitatory signs and at lethal levels a type of decerebrate rigidity. Phenytoin limits the repetitive firing of action potentials evoked by a sustained depolarization. This effect is mediated by a slowing of the rate of recovery of voltage-activated Na+ channels from inactivation, an action that is both voltage- (greater effect if membrane is depolarized) and use-dependent. At therapeutic concentrations, the effects on Na+ channels are selective, and no changes of spontaneous activity or responses to iontophoretically applied GABA or glutamate are detected. At concentrations 5- to 10-fold higher, multiple effects of phenytoin are evident, including reduction of spontaneous activity and enhancement of responses to GABA; these effects may underlie some of the unwanted toxicity associated with high levels of phenytoin.
PHARMACOKINETIC PROPERTIES. Phenytoin is available in 2 types of oral formulations that differ in their pharmacokinetics: rapid-release and extended-release forms. Once-daily dosing is possible only with the extended-release formulations, and due to differences in dissolution and other formulation-dependent factors, the plasma phenytoin level may change when converting from 1 formulation to another. Comparable doses can be approximated by considering “phenytoin equivalents,” but serum-level monitoring is necessary to assure therapeutic safety. Phenytoin is extensively bound (~90%) to serum proteins, mainly albumin. Increased proportions of free drug are evident in the neonate, in patients with hypoalbuminemia, and in uremic patients. Some agents can compete with phenytoin for binding sites on plasma proteins and increase free phenytoin. Valproate competes for protein binding sites and inhibits phenytoin metabolism, resulting in marked and sustained increases in free phenytoin. Measurement of free rather than total phenytoin permits direct assessment of this potential problem in patient management.
The plasma t1/2 of phenytoin (6 to 24 h at plasma concentrations < 10 μg/mL) increases with higher concentrations; as a result, plasma drug concentration increases disproportionately as dosage is increased, even with small adjustments for levels near the therapeutic range. The majority (95%) of phenytoin is metabolized by hepatic CYP2C9/10 and to a lesser extent CYP2C19 (see Table 21–3). Other drugs that are metabolized by these enzymes can inhibit the metabolism of phenytoin and increase its plasma concentration. Conversely, the degradation rate of other drugs that are substrates for these enzymes can be inhibited by phenytoin; one such drug is warfarin, and addition of phenytoin to a patient receiving warfarin can lead to bleeding disorders (see Chapter 30). Phenytoin has the capacity to induce CYPs (see Chapter 6); coadministration of phenytoin and medications metabolized by CYPs can lead to an increased degradation of such medications. For example, treatment with phenytoin can enhance the metabolism of oral contraceptives and lead to unplanned pregnancy. Phenytoin also has potential teratogenic effects. Carbamazepine, oxcarbazepine, phenobarbital, and primidone also can induce CYP3A4 and likewise might increase degradation of oral contraceptives.
The low water solubility of phenytoin hindered its intravenous use and led to production of fosphenytoin, a water-soluble prodrug. Fosphenytoin (CEREBYX, others) is converted into phenytoin by phosphatases in liver and red blood cells with a t1/2 of 8-15 min. Fosphenytoin is extensively bound (95-99%) to human plasma proteins, primarily albumin. Fosphenytoin is useful for adults with partial or generalized seizures when IV or intramuscular administration is indicated.
TOXICITY. The toxic effects of phenytoin depend on the route of administration, the duration of exposure, and the dosage. When fosphenytoin, the water-soluble prodrug, is administered intravenously at an excessive rate in the emergency treatment of status epilepticus, the most notable toxic signs are cardiac arrhythmias with or without hypotension, and/or CNS depression. Cardiac toxicity occurs more frequently in older patients and in those with known cardiac disease than young, healthy patients. These complications can be minimized by administering fosphenytoin at a rate of <150 mg of phenytoin sodium equivalents per minute. Acute oral overdosage results primarily in signs referable to the cerebellum and vestibular system; high doses have been associated with marked cerebellar atrophy. Toxic effects associated with chronic treatment also are primarily dose-related cerebellar-vestibular effects but also include other CNS effects, behavioral changes, increased frequency of seizures, GI symptoms, gingival hyperplasia, osteomalacia, and megaloblastic anemia. Hirsutism is an annoying untoward effect in young females. Usually, these phenomena can be diminished by adjustment of dosage. Serious adverse effects, including those on the skin, bone marrow, and liver, probably are manifestations of rare drug allergy and necessitate withdrawal of the drug. Moderate elevation of the plasma concentrations of hepatic transaminases sometimes are observed.
Gingival hyperplasia occurs in ~20% of all patients during chronic therapy. The condition can be minimized by good oral hygiene. Inhibition of release of antidiuretic hormone (ADH) has been observed. Hyperglycemia and glycosuria appear to be due to inhibition of insulin secretion. Osteomalacia, with hypocalcemia and elevated alkaline phosphatase activity, has been attributed to both altered metabolism of vitamin D and the attendant inhibition of intestinal absorption of Ca2+. Phenytoin also increases the metabolism of vitamin K and reduces the concentration of vitamin K–dependent proteins that are important for normal Ca2+ metabolism in bone. Hypersensitivity reactions include morbilliform rash in 2-5% of patients and occasionally more serious skin reactions, including Stevens-Johnson syndrome and toxic epidermal necrolysis. Hematological reactions include neutropenia and leukopenia. A few cases of red-cell aplasia, agranulocytosis, and mild thrombocytopenia also have been reported. Lymphadenopathy, resembling Hodgkin disease and malignant lymphoma, is associated with reduced immunoglobulin A (IgA) production. Hypoprothrombinemia and hemorrhage have occurred in the newborns of mothers who received phenytoin during pregnancy; vitamin K is effective treatment or prophylaxis.
PLASMA DRUG CONCENTRATIONS. A good correlation usually is observed between the total concentration of phenytoin in plasma and its clinical effect. Control of seizures generally is obtained with total concentrations > 10 μg/mL: toxic effects such as nystagmus develop at total concentrations ~20 μg/mL. Control of seizures generally is obtained with free phenytoin concentrations of 0.75-1.25 μg/mL.
DRUG INTERACTIONS. Drugs metabolized by CYP2C9 or CYP2C10 can increase the plasma concentration of phenytoin by decreasing its rate of metabolism. Carbamazepine, which may enhance the metabolism of phenytoin, causes a decrease in phenytoin concentration. Conversely, phenytoin reduces the concentration of carbamazepine.
THERAPEUTIC USES
Epilepsy. Phenytoin is one of the more widely used antiseizure agents, and it is effective against partial and tonic-clonic but not absence seizures. Phenytoin preparations differ significantly in bioavailability and rate of absorption. In general, patients should consistently be treated with the same drug from a single manufacturer. However, if it becomes necessary to switch between products, care should be taken to select a therapeutically equivalent product and patients should be monitored for loss of seizure control or onset of new toxicities.
Other Uses.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


