Pharmacotherapy of Inflammation, Fever, Pain, and Gout
The chapter describes the nonsteroidal anti-inflammatory drugs (NSAIDs) used to treat inflammation, pain, and fever and the drugs used for hyperuricemia and gout. The NSAIDS are first considered by class, then by groups of chemically similar agents described in more detail. Many of the basic properties of these drugs are summarized in Tables 34–2, 34–3, and 34–4. Most currently available traditional NSAIDs (tNSAIDs) act by inhibiting the prostaglandin (PG) G/H synthase enzymes, colloquially known as the cyclooxygenases (COXs; see Chapter 33). The inhibition of cyclooxygenase-2 (COX-2) is thought to mediate, in large part, the antipyretic, analgesic, and anti-inflammatory actions of tNSAIDs, while the simultaneous inhibition of cyclooxygenase-1 (COX-1) largely but not exclusively accounts for unwanted adverse effects in the GI tract. Selective inhibitors of COX-2 (celecoxib, etoricoxib, lumiracoxib) are a subclass of NSAIDs. Aspirin irreversibly acetylates COX; several structural subclasses of tNSAIDs, including propionic acid derivatives (ibuprofen, naproxen), acetic acid derivatives (indomethacin), and enolic acids (piroxicam) compete in a reversible manner with arachidonic acid (AA) at the active site of COX-1 and COX-2. Acetaminophen (paracetamol) is effective as an antipyretic and analgesic agent at typical doses that partly inhibit COXs, has only weak anti-inflammatory activity, and exhibits fewer GI side effects than the tNSAIDs.
INFLAMMATION, PAIN, AND FEVER
INFLAMMATION. The inflammatory process is the response to an injurious stimulus. It can be evoked by noxious agents, infections, antibodies, physical injuries. The ability to mount an inflammatory response is essential for survival in the face of environmental pathogens and injury; in some situations and diseases, the inflammatory response may be exaggerated and sustained without apparent benefit and even with severe adverse consequences. The inflammatory response is characterized mechanistically by:
• Transient local vasodilation and increased capillary permeability
• Infiltration of leukocytes and phagocytic cells
• Tissue degeneration and fibrosis
Many molecules are involved in the promotion and resolution of the inflammatory process. Histamine, bradykinin, 5-HT, prostanoids, leukotrienes (LTs), and platelet-activating factor are important mediators of inflammation (see Chapter 33).
Prostanoid biosynthesis is significantly increased in inflamed tissue. Inhibitors of the COXs, which depress prostanoid formation, are effective and widely used anti-inflammatory agents. Prostaglandin E2 (PGE2) and prostacyclin (PGI2) are the primary prostanoids that mediate inflammation. They increase local blood flow, vascular permeability, and leukocyte infiltration through activation of their respective receptors, EP2 and IP. PGD2, a major product of mast cells, contributes to inflammation in allergic responses, particularly in the lung.
Activation of endothelial cells plays a key role in “targeting” circulating cells to inflammatory sites. Endothelial activation results in leukocyte adhesion as the leukocytes recognize newly expressed L- and P-selectin and E-selectin with sialylated Lewis X and other glycoproteins on the leukocyte surface and endothelial intercellular adhesion molecule-1 (ICAM-1) with leukocyte integrins.
The recruitment of inflammatory cells to sites of injury also involves the concerted interactions of several types of soluble mediators. These include the complement factor C5a, PAF, and the eicosanoid LTB4 (see Chapter 33). All can act as chemotactic agonists. Several cytokines also play essential roles in orchestrating the inflammatory process, especially tumor necrosis factor (TNF) and interleukin-1 (IL-1). Other cytokines and growth factors (e.g., IL-2, IL-6, IL-8, GM-CSF) contribute to manifestations of the inflammatory response. The concentrations of many of these factors are increased in the synovia of patients with inflammatory arthritis. Glucocorticoids interfere with the synthesis and actions of cytokines, such as IL-1 or TNF-t (see Chapter 35). Although some of the actions of these cytokines are accompanied by the release of PGs and thromboxane A2 (TxA2), cyclooxygenase (COX) inhibitors appear to block only their pyrogenic effects.
PAIN. Inflammatory mediators released from nonneuronal cells during tissue injury increase the sensitivity of nociceptors and potentiate pain perception. Among these mediators are bradykinin, H+, 5-HT, ATP, neurotrophins (nerve growth factor), LTs, and PGs. PGE2 and PGI2 reduce the threshold to stimulation of nociceptors, causing peripheral sensitization. Centrally active PGE2 and perhaps also PGD2, PGI2, and PGF2α contribute to central sensitization, an increase in excitability of spinal dorsal horn neurons that causes hyperalgesia and allodynia in part by disinhibition of glycinergic pathways.
FEVER. The hypothalamus regulates the set point at which body temperature is maintained. This set point is elevated in fever, reflecting an infection, or resulting from tissue damage, inflammation, graft rejection, or malignancy. These conditions all enhance formation of cytokines such as IL-1 β, IL-6, TNF-α, and interferons, which act as endogenous pyrogens. The initial phase of the thermoregulatory response to such pyrogens may be mediated by ceramide release in neurons of the preoptic area in the anterior hypothalamus. A late response is mediated by coordinate induction of COX-2 and formation of PGE2. PGE2 can cross the blood-brain barrier and acts on EP3 and perhaps EP1 receptors on thermosensitive neurons. This triggers the hypothalamus to elevate body temperature by promoting an increase in heat generation and a decrease in heat loss. NSAIDs suppress this response by inhibiting PGE2 synthesis.
NONSTEROIDAL ANTI-INFLAMMATORY DRUGS
NSAIDs are classified as tNSAIDs, which inhibit both COX-1 and COX-2, and COX-2–selective NSAIDs. Most NSAIDs are competitive, reversible, active site inhibitors of the COX enzymes. However, aspirin (acetyl salicylic acid, ASA) acetylates the isozymes and inhibits them irreversibly; thus, aspirin often is distinguished from the tNSAIDs. Similarly, acetaminophen, which is antipyretic and analgesic but largely devoid of anti-inflammatory activity, also is conventionally segregated from the group.
The vast majority of tNSAID compounds are organic acids with relatively low pKa values (Figure 34–1). Even the nonacidic parent drug nabumetone is converted to an active acetic acid derivative in vivo. As organic acids, the compounds generally are well absorbed orally, highly bound to plasma proteins, and excreted either by glomerular filtration or by tubular secretion. They also accumulate in sites of inflammation, where the pH is lower, potentially confounding the relationship between plasma concentrations and duration of drug effect. Most COX-2–selective NSAIDs are diaryl heterocyclic compounds with a relatively bulky side group, which aligns with a large side pocket in the AA binding channel of COX-2 but hinders its optimal orientation in the smaller binding channel of COX-1. Both tNSAIDs and the COX-2–selective NSAIDs generally are hydrophobic drugs, a feature that allows them to access the hydrophobic arachidonate binding channel and results in shared pharmacokinetic characteristics. Again, aspirin and acetaminophen are exceptions to this rule.
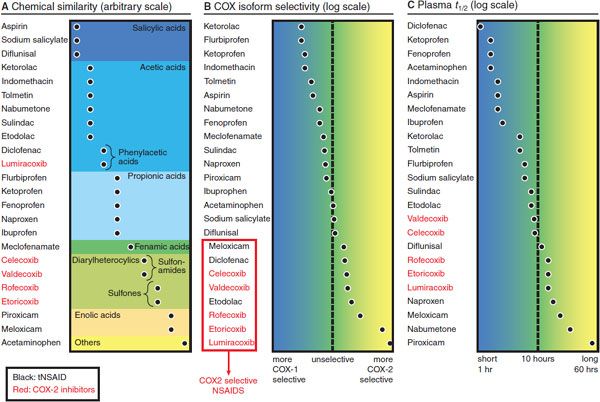
Figure 34–1 Classification of NSAIDs by chemical similarity (panel A), cyclooxygenase (COX) isoform selectivity (panel B), and plasma t1/2 (panel C). The COX selectivity chart is plotted from data published in Warner et al., 1999, and FitzGerald and Patrono, 2001. tNSAIDs, traditional nonsteroidal anti-inflammatory drugs.
MECHANISM OF ACTION
CYCLOOXYGENASE INHIBITION. The principal therapeutic effects of NSAIDs derive from their ability to inhibit PG production. The first enzyme in the PG synthetic pathway is COX, also known as PG G/H synthase. This enzyme converts AA to the unstable intermediates PGG2 and PGH2 and leads to the production of the prostanoids, TxA2, and a variety of PGs (see Chapter 33). There are 2 forms of COX, COX-1 and COX-2. COX-1, expressed constitutively in most cells, is the dominant source of prostanoids for housekeeping functions. Conversely, COX-2, induced by cytokines, shear stress, and tumor promoters, is the more important source of prostanoid formation in inflammation and perhaps in cancer (see Chapter 33). COX-1 is the dominant isoform in gastric epithelial cells and is thought to be the major source of cytoprotective PG formation. Inhibition of COX-1 accounts for the gastric adverse events that complicate therapy with tNSAIDs.
Aspirin and NSAIDs inhibit the COX enzymes and PG production; they do not inhibit the lipoxygenase (LOX) pathways of AA metabolism and hence do not suppress LT formation (see Chapter 33).
IRREVERSIBLE CYCLOOXYGENASE INHIBITION BY ASPIRIN. Aspirin covalently modifies COX-1 and COX-2, irreversibly inhibiting COX activity. This is an important distinction from all the NSAIDs because the duration of aspirin’s effects is related to the turnover rate of COXs in different target tissues.
The importance of enzyme turnover in recovery from aspirin action is most notable in platelets, which, being anucleate, have a markedly limited capacity for protein synthesis. Thus, the consequences of inhibition of platelet COX-1 last for the lifetime of the platelet. Inhibition of platelet COX-1–dependent TxA2 formation therefore is cumulative with repeated doses of aspirin (at least as low as 30 mg/day) and takes ~8-12 days (the platelet turnover time) to recover fully once therapy has been stopped. The unique sensitivity of platelets to inhibition by such low doses of aspirin is related to their presystemic inhibition in the portal circulation before aspirin is deacetylated to salicylate on first pass through the liver. In contrast to aspirin, salicylic acid has no acetylating capacity. It is a weak, reversible, competitive inhibitor of COX.
SELECTIVE INHIBITION OF CYCLOOXYGENASE-2. The therapeutic use of the tNSAIDs is limited by their poor GI tolerability. Since COX-1 was the predominant source of cytoprotective PGs formed by the GI epithelium, selective inhibitors of COX-2 were developed to afford efficacy similar to tNSAIDs with better GI tolerability. Six COX-2 inhibitors, the coxibs, were initially approved for use: celecoxib, rofecoxib, valdecoxib and its prodrug parecoxib, etoricoxib, and lumiracoxib. Most coxibs have been either restricted in their use or withdrawn from the market in view of their adverse event profile. Celecoxib (CELEBREX) currently is the only COX-2 inhibitor licensed for use in the U.S.
ADME
NSAIDs are rapidly absorbed following oral ingestion, and peak plasma concentrations are reached within 2-3 h. Food intake may delay absorption and systemic availability (i.e., fenoprofen, sulindac). Antacids, commonly prescribed to patients on NSAID therapy, variably delay, absorption. Some compounds (e.g., diclofenac, nabumetone) undergo first-pass or presystemic elimination. Aspirin begins to acetylate platelets within minutes of reaching the presystemic circulation.
Most NSAIDs are extensively bound to plasma proteins (95-99%), usually albumin. Highly protein bound NSAIDs have the potential to displace other drugs, if they compete for the same binding sites. Most NSAIDs are distributed widely throughout the body and readily penetrate arthritic joints, yielding synovial fluid concentrations in the range of half the plasma concentration (i.e., ibuprofen, naproxen, piroxicam). Most NSAIDs achieve sufficient concentrations in the CNS to have a central analgesic effect. Celecoxib is particularly lipophilic and moves readily into the CNS. Lumiracoxib is more acidic than other COX-2–selective NSAIDs, which may favor its accumulation at sites of inflammation.
Plasma t1/2 varies considerably among NSAIDs. Ibuprofen, diclofenac, and acetaminophen have t1/2 of 1-4 h, while piroxicam has a t1/2 of 50 h at steady state. The t1/2 of COX-2–selective NSAIDs vary (2-6 h for lumiracoxib, 6-12 h for celecoxib, and 20-26 h for etoricoxib). Hepatic biotransformation and renal excretion are the principal routes of metabolism and elimination of the majority of NSAIDs. Acetaminophen, at therapeutic doses, is oxidized only to a small degree to form traces of the highly reactive metabolite, N-acetyl-p-benzoquinone imine (NAPQI). Following overdose (usually >10 g of acetaminophen), however, the principal metabolic pathways are saturated, and hepatotoxic NAPQI concentrations can be formed (see Figure 4–5). Rarely, other NSAIDs also may be complicated by hepatotoxicity (e.g., diclofenac, lumiracoxib). NSAIDs usually are not removed by hemodialysis due to their extensive plasma protein binding; salicylic acid is an exemption to this rule. NSAIDs are not recommended in advanced hepatic or renal disease.
THERAPEUTIC USES
All NSAIDs are antipyretic, analgesic, and anti-inflammatory, with the exception of acetaminophen, which is antipyretic and analgesic but is largely devoid of anti-inflammatory activity.
INFLAMMATION. NSAIDs provide mostly symptomatic relief from pain and inflammation associated with musculoskeletal disorders, such as rheumatoid arthritis and osteoarthritis. Some NSAIDs are approved for the treatment of ankylosing spondylitis and gout.
PAIN. NSAIDs are effective only against pain of low to moderate intensity. Although their maximal efficacy is much less than the opioids, NSAIDs lack the unwanted adverse effects of opiates. Coadministration of NSAIDs can reduce the opioid dose needed for sufficient pain control and reduce the likelihood of adverse opioid effects. NSAIDs are particularly effective when inflammation has caused peripheral and/or central sensitization of pain perception. An exception to this is menstrual pain. The release of PGs by the endometrium during menstruation may cause severe cramps and other symptoms of primary dysmenorrhea; treatment of this condition with NSAIDs has met with considerable success. NSAIDs are commonly used to treat migraine attacks and can be combined with drugs such as the triptans (e.g., TREXIMET, a fixed-dose combination of naproxen and sumatriptan) or with antiemetics to aid relief of the associated nausea. NSAIDs lack efficacy in neuropathic pain.
FEVER. Antipyretic therapy is reserved for patients in whom fever in itself may be deleterious and for those who experience considerable relief when fever is lowered. NSAIDs reduce fever in most situations, but not the circadian variation in temperature or the rise in response to exercise or increased ambient temperature. COX-2 is the dominant source of PGs that mediate the rise in temperature evoked by bacterial lipopolysaccharide (LPS) administration.
FETAL CIRCULATORY SYSTEM. PGs are implicated in the maintenance of patency of the ductus arteriosus, and indomethacin, ibuprofen, and other tNSAIDs have been used in neonates to close the inappropriately patent ductus.
CARDIOPROTECTION. Ingestion of aspirin prolongs bleeding time. This effect is due to irreversible acetylation of platelet COX and the consequent inhibition of platelet function. It is the permanent suppression of platelet TxA2 formation that is thought to underlie the cardioprotective effect of aspirin.
Aspirin reduces the risk of serious vascular events in high-risk patients (e.g., those with previous myocardial infarction) by 20-25%. Low-dose (<100 mg/day) aspirin is relatively selective for COX-1 and is associated with a lower risk for GI adverse events. However, low-dose aspirin increases the incidence of serious GI bleeds. It also increases the incidence of intracranial bleeds. The benefit from aspirin outweighs these risks in the case of secondary prevention of cardiovascular disease. Given their relatively short t1/2 and reversible COX inhibition, most other tNSAIDs are not thought to afford cardioprotection. Data suggest that cardioprotection is lost when combining low-dose aspirin with ibuprofen. COX-2–selective NSAIDs are devoid of antiplatelet activity, as mature platelets do not express COX-2.
OTHER CLINICAL USES
Systemic Mastocytosis. Systemic mastocytosis is a condition in which there are excessive mast cells in the bone marrow, reticuloendothelial system, GI system, bones, and skin. In patients with systemic mastocytosis, PGD2, released from mast cells is the major mediator of severe episodes of flushing, vasodilation, and hypotension. The addition of aspirin or ketoprofen has provided relief. However, aspirin and tNSAIDs can cause degranulation of mast cells, so blockade with H1 and H2 histamine receptor antagonists should be established before NSAIDs are initiated.
Niacin Tolerability. Large doses of niacin (nicotinic acid) effectively lower serum cholesterol levels, reduce low-density lipoprotein, and raise high-density lipoprotein (see Chapter 31). However, niacin induces intense facial flushing mediated largely by release of PGD2 from the skin, which can be inhibited by treatment with aspirin.
Cancer Chemoprevention. Epidemiological studies suggested that frequent use of aspirin is associated with as much as a 50% decrease in the risk of colon cancer. Similar observations have been made with NSAID use in this and other cancers.
ADVERSE EFFECTS OF NSAID THERAPY
Common adverse events of aspirin and NSAIDs are outlined in Table 34–1.
Table 34–1
Common and Shared Side Effects of NSAIDs

GASTROINTESTINAL. The most common symptoms associated with these drugs are GI, including anorexia, nausea, dyspepsia, abdominal pain, and diarrhea. These symptoms may be related to the induction of gastric or intestinal ulcers, which is estimated to occur in 15-30% of regular users. Ulceration may be complicated by bleeding, perforation, or obstruction. The risk is further increased in those with Helicobacter pylori infection, heavy alcohol consumption, or other risk factors for mucosal injury, including the concurrent use of glucocorticoids. All selective COX-2 inhibitors are less prone to induce gastric ulcers than equally efficacious doses of tNSAIDs.
CARDIOVASCULAR. COX-2–selective NSAIDs were developed to improve the GI safety. However, clinical trials—with celecoxib, valdecoxib (withdrawn), and rofecoxib (withdrawn)—revealed an increase in the incidence of myocardial infarction, stroke, and thrombosis. COX-2-inhibitors depress formation of PGI2 but do not inhibit COX-1 catalyzed formation of platelet thromboxane TxA2. PGI2 inhibits platelet aggregation and constrains the effect of prothrombotic and atherogenic stimuli by TxA2.
BLOOD PRESSURE, RENAL, AND RENOVASCULAR ADVERSE EVENTS. NSAIDs and COX-2 inhibitors have been associated with renal and renovascular adverse events. In patients with congestive heart failure, hepatic cirrhosis, chronic kidney disease, hypovolemia, and other states of activation of the sympathoadrenal or renin–angiotensin systems, PG formation becomes crucial. NSAIDs are associated with loss of the PG-induced inhibition of both the reabsorption of Cl– and the action of antidiuretic hormone, leading to the retention of salt and water. Epidemiological studies suggest hypertensive complications occur more commonly in patients treated with coxibs than with tNSAIDs.
ANALGESIC NEPHROPATHY. Analgesic nephropathy is a condition of slowly progressive renal failure, decreased concentrating capacity of the renal tubule, and sterile pyuria. Risk factors are the chronic use of high doses of combinations of NSAIDs and frequent urinary tract infections.
PREGNANCY AND LACTATION. Myometrial COX-2 expression and levels of PGE2 and PGF2α increase markedly in the myometrium during labor. Prolongation of gestation by NSAIDs has been demonstrated in humans. Some NSAIDs, particularly indomethacin, have been used off-label to terminate preterm labor. However, this use is associated with closure of the ductus arteriosus and impaired fetal circulation in utero, particularly in fetuses older than 32 weeks’ gestation. COX-2–selective inhibitors have been used off-label as tocolytic agents; this use has been associated with stenosis of the ductus arteriosus and oligohydramnios. Finally, the use of NSAIDs and aspirin late in pregnancy may increase the risk of postpartum hemorrhage. Therefore, pregnancy, especially close to term, is a relative contraindication to the use of all NSAIDs. In addition, their use must be weighed against potential fetal risk, even in cases of premature labor, and especially in cases of pregnancy-induced hypertension.
HYPERSENSITIVITY. Hypersensitivity symptoms to aspirin and NSAIDs range from vasomotor rhinitis, generalized urticaria, and bronchial asthma to laryngeal edema, bronchoconstriction, flushing, hypotension, and shock. Aspirin intolerance is a contraindication to therapy with any other NSAID because cross-sensitivity. Treatment of aspirin hypersensitivity is similar to that of other severe hypersensitivity reactions, with support of vital organ function and administration of epinephrine.
ASPIRIN RESISTANCE. All forms of treatment failure with aspirin have been collectively called aspirin resistance. Genetic variants of COX-1 that cosegregate with resistance have been described, but the relation to clinical outcome is not clear.
REYE SYNDROME. Due to the possible association with Reye syndrome, aspirin and other salicylates are contraindicated in children and young adults < 20 years of age with viral illness–associated fever. Reye syndrome, a severe and often fatal disease, is characterized by the acute onset of encephalopathy, liver dysfunction, and fatty infiltration of the liver and other viscera. Although a mechanistic understanding is lacking, the epidemiologic association between aspirin use and Reye syndrome is sufficiently strong that aspirin and bismuth subsalicylate labels now must indicate the risk. As the use of aspirin in children has declined dramatically, so has the incidence of Reye syndrome. Acetaminophen has not been implicated in Reye syndrome and is the drug of choice for antipyresis in children and young adults.
CONCOMITANT NSAIDS AND LOW-DOSE ASPIRIN. Many patients combine either tNSAIDs or COX-2 inhibitors with low-dose aspirin for “cardioprotection.” Epidemiological studies suggest that this combination therapy increases significantly the likelihood of GI adverse events over either class of NSAID alone.
DRUG INTERACTIONS
Angiotensin-converting enzyme (ACE) inhibitors act, at least partly, by preventing the breakdown of kinins that stimulate PG production (see Figure 32–2). Thus, it is logical that NSAIDs might attenuate the effectiveness of ACE inhibitors by blocking the production of vasodilator and natriuretic PGs. Due to hyperkalemia, the combination of NSAIDs and ACE inhibitors also can produce marked bradycardia leading to syncope, especially in the elderly and in patients with hypertension, diabetes mellitus, or ischemic heart disease. Corticosteroids and SSRIs may increase the frequency or severity of GI complications when combined with NSAIDs. NSAIDs may augment the risk of bleeding in patients receiving warfarin both because almost all tNSAIDs suppress normal platelet function temporarily during the dosing interval and because some NSAIDs also increase warfarin levels by interfering with its metabolism. Many NSAIDs are highly bound to plasma proteins and thus may displace other drugs from their binding sites. Such interactions can occur in patients given salicylates or other NSAIDs together with warfarin, sulfonylurea hypoglycemic agents, or methotrexate; the dosage of such agents may require adjustment to prevent toxicity. Patients taking lithium should be monitored because certain NSAIDs (e.g., piroxicam) can reduce the renal excretion of this drug and lead to toxicity, while others can decrease lithium levels (e.g., sulindac).
PEDIATRIC AND GERIATRIC USE
THERAPEUTIC USES IN CHILDREN. Therapeutic indications for NSAID use in children include fever, mild pain, postoperative pain, and inflammatory disorders, such as juvenile arthritis and Kawasaki disease. Only drugs that have been extensively tested in children should be used (acetaminophen, ibuprofen, and naproxen).
PHARMACOKINETICS IN CHILDREN. NSAID dosing recommendations frequently are based on extrapolation of pharmacokinetic data from adults or children >2 years, and there is often insufficient data for dose selection in younger infants. For example, the pharmacokinetics of the most commonly used NSAID in children, acetaminophen, differ substantially between the neonatal period and older children or adults. The systemic bioavailability of rectal acetaminophen formulations in neonates and preterm babies is higher than in older patients. Acetaminophen clearance is reduced in preterm neonates probably due to their immature glucuronide conjugation system (sulfation is the principal route of biotransformation at this age). Therefore, acetaminophen dosing intervals need to be extended (8-12 h) or daily doses reduced to avoid accumulation and liver toxicity. Aspirin elimination also is delayed in neonates and young infants compared to adults bearing the risk of accumulation. Disease also may affect NSAID disposition in children. For example, ibuprofen plasma concentrations are reduced and clearance increased (~80%) in children with cystic fibrosis. This probably is related to the GI and hepatic pathologies associated with this disease. Aspirin’s kinetics are markedly altered during the febrile phase of rheumatic fever or Kawasaki vasculitis. The reduction in serum albumin associated with these conditions causes an elevation of the free salicylate concentration, which may saturate renal excretion and result in salicylate accumulation to toxic levels. In addition to dose reduction, monitoring of the free drug may be warranted in these situations.
PHARMACOKINETICS IN THE ELDERLY. The clearance of many NSAIDs is reduced in the elderly due to changes in hepatic metabolism. NSAIDs with a long t1/2 and primarily oxidative metabolism (i.e., piroxicam, tenoxicam, celecoxib) have elevated plasma concentrations in elderly patients. For example, plasma concentrations after the same dose of celecoxib may rise up to 2-fold higher in patients >65 years than in patients <50 years of age, warranting dose adjustment. The capacity of plasma albumin to bind drugs is diminished in older patients and may result in higher concentrations of unbound NSAIDs. The higher susceptibility of older patients to GI complications may be due to a reduction in gastric mucosal defense and to elevated total and/or free NSAID concentrations. Generally, it is advisable to start most NSAIDs at a low dosage in the elderly and increase the dosage only if the therapeutic efficacy is insufficient.
SPECIFIC PROPERTIES OF INDIVIDUAL NSAIDS
General properties shared by NSAIDs were considered in the preceding section, “Nonsteroidal Anti-Inflammatory Drugs.” In this section, important characteristics of individual substances are discussed. NSAIDs are grouped by their chemical similarity, as in Figure 34–1.
ASPIRIN AND OTHER SALICYLATES
The salicylates include, aspirin, salicylic acid, methyl salicylate, diflunisal, salsalate, olsalazine, and sulfasalazine. Aspirin is the most widely consumed analgesic, antipyretic, and anti-inflammatory agent. Because aspirin is so available, the possibility of misuse and serious toxicity is underappreciated.
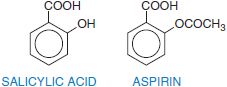
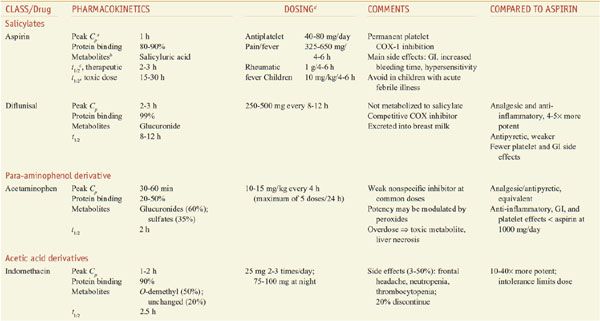
Salicylic acid is so irritating that it can only be used externally; therefore various derivatives of this acid have been synthesized for systemic use. For example, aspirin is the acetate ester of salicylic acid. Table 34–2 summarizes the clinical pharmacokinetic properties of 2 salicylates, aspirin and diflunisal.
Table 34–2
Comparison of NSAIDS: Salicylates, Acetaminophen, and Acetic Acid Derivatives

MECHANISM OF ACTION
Salicylates generally act by virtue of their content of salicylic acid. The effects of aspirin are largely caused by its capacity to acetylate proteins, as described in “Irreversible Cyclooxygenase Inhibition by Aspirin,” above.
ADME
ABSORPTION. Orally ingested salicylates are absorbed rapidly, partly from the stomach but mostly from the upper small intestine. Appreciable concentrations are found in plasma in <30 min; after a single dose, the peak plasma level is reached in ~1 h and then declines gradually. The rate of absorption is determined by disintegration and dissolution rates of the tablets administered, the pH at the mucosal surface, and gastric emptying time. The presence of food delays absorption of salicylates. Rectal absorption of salicylate usually is slower than oral absorption and is incomplete and inconsistent.
Salicylic acid is absorbed rapidly from the intact skin, especially when applied in oily liniments or ointments, and systemic poisoning has occurred from its application to large areas of skin. Methyl salicylate likewise is speedily absorbed when applied cutaneously; however, its GI absorption may be delayed many hours, making gastric lavage effective for removal even in poisonings that present late after oral ingestion.
DISTRIBUTION. After absorption, salicylates are distributed throughout most body tissues and transcellular fluids, primarily by pH-dependent passive processes. Salicylates are transported actively out of the CSF across the choroid plexus. The drugs readily cross the placental barrier. Ingested aspirin mainly is absorbed as such, but some enters the systemic circulation as salicylic acid after hydrolysis by esterases in the GI mucosa and liver. Roughly 80-90% of the salicylate in plasma is bound to proteins, especially albumin; the proportion of the total that is bound declines as plasma concentrations increase. Hypoalbuminemia, as may occur in rheumatoid arthritis, is associated with a proportionately higher level of free salicylate in the plasma. Salicylate competes with a variety of compounds for plasma protein binding sites; these include thyroxine, triiodothyronine, penicillin, phenytoin, sulfinpyrazone, bilirubin, uric acid, and other NSAIDs such as naproxen. Aspirin is bound to a more limited extent; however, it acetylates human plasma albumin in vivo by reaction with the ε-amino group of lysine and may change the binding of other drugs to albumin. Aspirin also acetylates hormones, DNA, and hemoglobin and other proteins.
METABOLISM AND ELIMINATION. The 3 chief metabolic products are salicyluric acid (the glycine conjugate), the ether or phenolic glucuronide, and the ester or acyl glucuronide. Salicylates and their metabolites are excreted in the urine The excretion of free salicylates is variable and depends on the dose and the urinary pH. For example, the clearance of salicylate is about 4 times as great at pH 8 as at pH 6, and it is well above the glomerular filtration rate at pH 8. High rates of urine flow decrease tubular reabsorption, whereas the opposite is true in oliguria. The plasma t1/2 for aspirin is ~20 min, and for salicylate is 2-3 h at antiplatelet doses, rising to 12 h at usual anti-inflammatory doses. The t1/2 of salicylate may rise to 15-30 h at high therapeutic doses or when there is intoxication. This dose-dependent elimination is the result of the limited capacity of the liver to form salicyluric acid and the phenolic glucuronide, resulting in a larger proportion of unchanged drug being excreted in the urine at higher doses. Salicylate clearance is reduced and salicylate exposure is significantly increased in the elderly. The plasma concentration of salicylate is increased by conditions that decrease glomerular filtration rate or reduce proximal tubule secretion, such as renal disease or the presence of inhibitors that compete for the transport system (e.g., probenecid).
THERAPEUTIC USES
SYSTEMIC USES. The analgesic–antipyretic dose of aspirin for adults is 324-1000 mg orally every 4-6 h. The anti-inflammatory doses of aspirin recommended for arthritis, spondyloarthropathies, and systemic lupus erythematosus
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


