Pharmacotherapy of Gastric Acidity, Peptic Ulcers, and Gastroesophageal Reflux Disease
The treatment and prevention of acid-related disorders are accomplished by decreasing gastric acidity and enhancing mucosal defense. The appreciation that an infectious agent, Helicobacter pylori, plays a key role in the pathogenesis of acid-peptic diseases has stimulated new approaches to prevention and therapy.
PHYSIOLOGY OF GASTRIC SECRETION
Gastric acid secretion is a complex and continuous process: neuronal (acetylcholine, ACh), paracrine (histamine), and endocrine (gastrin) factors all regulate the secretion of H+ by parietal cells (Figure 45–1).
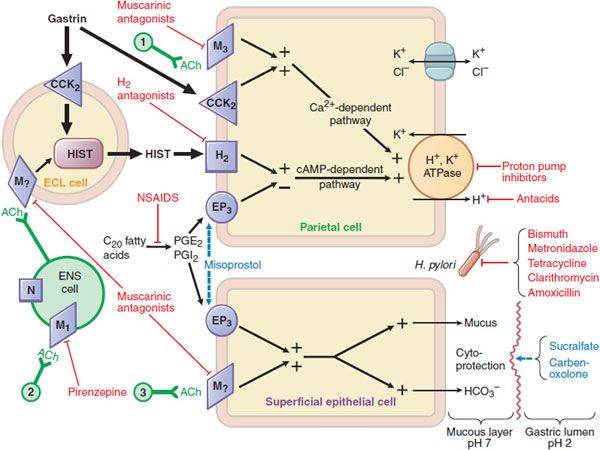
Figure 45–1 Pharmacologist’s view of gastric secretion and its regulation: the basis for therapy of acid-peptic disorders. Shown are the interactions among an enterochromaffin-like (ECL) cell that secretes histamine, a ganglion cell of the enteric nervous system (ENS), a parietal cell that secretes acid, and a superficial epithelial cell that secretes mucus and bicarbonate. Physiological pathways, shown in solid black, may be stimulatory (+) or inhibitory (–). 1 and 3 indicate possible inputs from postganglionic cholinergic fibers; 2 shows neural input from the vagus nerve. Physiological agonists and their respective membrane receptors include acetylcholine (ACh), muscarinic (M), and nicotinic (N) receptors; gastrin, cholecystokinin receptor 2 (CCK2); histamine (HIST), H2 receptor; and prostaglandin E2 (PGE2), EP3 receptor. A red indicates targets of pharmacological antagonism. A light blue dashed arrow indicates a drug action that mimics or enhances a physiological pathway. Shown in red are drugs used to treat acid-peptic disorders. NSAIDs are nonsteroidal anti-inflammatory drugs, which can induce ulcers via inhibition of cyclooxygenase.
Specific receptors (M3, H2, and CCK2, respectively) are on the basolateral membrane of parietal cells in the body and fundus of the stomach. Some of these receptors are also present on enterochromaffin-like (ECL) cells, where they regulate the release of histamine. The H2 receptor is a GPCR that activates the Gs–adenylyl cyclase–cyclic AMP–PKA pathway. ACh and gastrin signal through GPCRs that couple to the Gq–PLC-IP3–Ca2+ pathway in parietal cells. In parietal cells, the cyclic AMP and the Ca2+-dependent pathways activate H+, K+-ATPase (the proton pump), which exchanges H+ and K+ across the parietal cell membrane. This pump generates the largest ion gradient known in vertebrates, with an intracellular pH of ~7.3 and an intracanalicular pH of ~0.8.
ACh release from postganglionic vagal fibers directly stimulates gastric acid secretion through muscarinic M3 receptors on the basolateral membrane of parietal cells. The CNS predominantly modulates the activity of the enteric nervous system via ACh, stimulating gastric acid secretion in response to the sight, smell, taste, or anticipation of food (the “cephalic” phase of acid secretion). ACh also indirectly affects parietal cells by increasing the release of histamine from the ECL cells and of gastrin from G cells. ECL cells, the source of gastric histamine, usually are in close proximity to parietal cells. Histamine acts as a paracrine mediator, diffusing from its site of release to nearby parietal cells, where it activates H2 receptors to stimulate gastric acid secretion.
Gastrin, produced by antral G cells, is the most potent inducer of acid secretion. Multiple pathways stimulate gastrin release, including CNS activation, local distention, and chemical components of the gastric contents. Gastrin stimulates acid secretion indirectly by inducing the release of histamine by ECL cells; a direct effect on parietal cells also plays a lesser role. Somatostatin (SST), which is produced by antral D cells, inhibits gastric acid secretion. Acidification of the gastric luminal pH to <3 stimulates SST release, which in turn suppresses gastrin release in a negative feedback loop. SST-producing cells are decreased in patients with H. pylori infection, and the consequent reduction of SST’s inhibitory effect may contribute to excess gastrin production.
GASTRIC DEFENSES AGAINST ACID. The extremely high concentration of H+ in the gastric lumen requires robust defense mechanisms to protect the esophagus and the stomach. The primary esophageal defense is the lower esophageal sphincter, which prevents reflux of acidic gastric contents into the esophagus. The stomach protects itself from acid damage by a number of mechanisms that require adequate mucosal blood flow. One key defense is the secretion of a mucus layer that helps to protect gastric epithelial cells by trapping secreted bicarbonate at the cell surface. Gastric mucus is soluble when secreted but quickly forms an insoluble gel that coats the mucosal surface of the stomach, slows ion diffusion, and prevents mucosal damage by macromolecules such as pepsin. Mucus production is stimulated by prostaglandins E2 and I2, which also directly inhibit gastric acid secretion by parietal cells. Thus, drugs that inhibit prostaglandin formation (e.g., NSAIDs, ethanol) decrease mucus secretion and predispose to the development of acid-peptic disease. Figure 45–1 outlines the rationale and pharmacological basis for the therapy of acid-peptic diseases. The proton pump inhibitors are used most commonly, followed by the histamine H2 receptor antagonists.
PROTON PUMP INHIBITORS
The most potent suppressors of gastric acid secretion are inhibitors of the gastric H+, K+-ATPase (proton pump) (Figure 45–2). These drugs diminish the daily production of acid (basal and stimulated) by 80-95%.
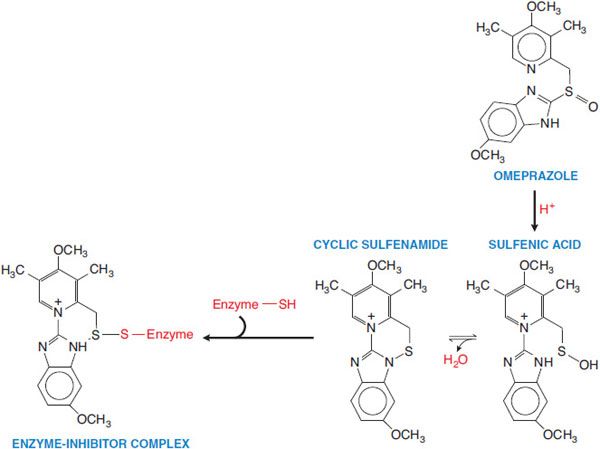
Figure 45–2 Activation of prodrug proton pump inhibitor. Omeprazole is converted to a sulfenamide in the acidic secretory canaliculi of the parietal cell. The sulfenamide interacts covalently with sulfhydryl groups in the proton pump, thereby irreversibly inhibiting its activity. Lansoprazole, rabeprazole, and pantoprazole undergo analogous conversions.
CHEMISTRY; MECHANISM OF ACTION; PHARMACOLOGY. Six proton pump inhibitors are available for clinical use: omeprazole (PRILOSEC, others) and its S-isomer, esomeprazole (NEXIUM), lansoprazole (PREVACID) and its R-enantiomer, dexlansoprazole (KAPIDEX), rabeprazole (ACIPHEX), and pantoprazole (PROTONIX, others). All proton pump inhibitors have equivalent efficacy at comparable doses.
Proton pump inhibitors (PPIs) are prodrugs that require activation in an acid environment. After absorption into the systemic circulation, the prodrug diffuses into the parietal cells of the stomach and accumulates in the acidic secretory canaliculi. Here, it is activated by proton-catalyzed formation of a tetracyclic sulfenamide (see Figure 45–2), trapping the drug so that it cannot diffuse back across the canalicular membrane. The activated form then binds covalently with sulfhydryl groups of cysteines in the H+, K+-ATPase, irreversibly inactivating the pump molecule. Acid secretion resumes only after new pump molecules are synthesized and inserted into the luminal membrane, providing a prolonged (up to 24- to 48-h) suppression of acid secretion, despite the much shorter plasma t1/2 ~ 0.5-2 h of the parent compounds.
To prevent degradation of PPIs by acid in the gastric lumen and improve oral bioavailability, oral dosage forms are supplied in different formulations:
• Enteric-coated drugs contained inside gelatin capsules (omeprazole, dexlansoprazole, esomeprazole, and lansoprazole)
• Enteric-coated granules supplied as a powder for suspension (lansoprazole)
• Enteric-coated tablets (pantoprazole, rabeprazole, and omeprazole)
• Powdered omeprazole combined with sodium bicarbonate (ZEGERID) contained in capsules and formulated for oral suspension
Patients for whom the oral route of administration is not available can be treated parenterally with esomeprazole, pantoprazole, or lansoprazole. The FDA-approved dose of intravenous pantoprazole for gastroesophageal reflux disease is 40 mg daily for up to 10 days. Higher doses (e.g., 160-240 mg in divided doses) are used to manage hypersecretory conditions such as the Zollinger-Ellison syndrome.
ADME. Because an acidic pH in the parietal cell acid canaliculi is required for drug activation and food stimulates acid production, these drugs ideally should be given ~30 min before meals. Concurrent administration of food may reduce somewhat the rate of absorption of PPIs. Once in the small bowel, PPIs are rapidly absorbed, highly protein bound, and extensively metabolized by hepatic CYPs, particularly CYP2C19 and CYP3A4. Asians are more likely than whites or African Americans to have the CYP2C19 genotype that correlates with slow metabolism of PPIs (23% vs. 3%, respectively), which may contribute to heightened efficacy and/or toxicity in this ethnic group.
Because not all pumps and all parietal cells are active simultaneously, maximal suppression of acid secretion requires several doses of the PPIs. For example, it may take 2-5 days of therapy with once-daily dosing to achieve the 70% inhibition of proton pumps that is seen at steady state. More frequent initial dosing (e.g., twice daily) will reduce the time to achieve full inhibition but is not proven to improve patient outcome. The resulting proton pump inhibition is irreversible; thus, acid secretion is suppressed for 24-48 h, or more, until new proton pumps are synthesized and incorporated into the luminal membrane of parietal cells. Chronic renal failure does not lead to drug accumulation with once-a-day dosing of the PPIs. Hepatic disease substantially reduces the clearance of esomeprazole and lansoprazole.
ADVERSE EFFECTS AND DRUG INTERACTIONS. PPIs generally cause remarkably few adverse effects. The most common side effects are nausea, abdominal pain, constipation, flatulence, and diarrhea. Subacute myopathy, arthralgias, headaches, and skin rashes also have been reported. As noted earlier, PPIs are metabolized by hepatic CYPs and therefore may interfere with the elimination of other drugs cleared by this route. PPIs have been observed to interact with warfarin (esomeprazole, lansoprazole, omeprazole, and rabeprazole), diazepam (esomeprazole and omeprazole), and cyclosporine (omeprazole and rabeprazole). Among the PPIs, only omeprazole inhibits CYP2C19 (thereby decreasing the clearance of disulfiram, phenytoin, and other drugs) and induces the expression of CYP1A2 (thereby increasing the clearance of imipramine, several antipsychotic drugs, tacrine, and theophylline). There is emerging evidence that omeprazole can inhibit conversion of clopidogrel (at the level of CYP2C19) to the active anticoagulating form. Pantoprazole is less likely to result in this interaction; concurrent use of clopidogrel and PPIs (mainly pantoprazole) significantly reduces GI bleeding without increasing adverse cardiac events (see Chapter 30).
Chronic treatment with omeprazole decreases the absorption of vitamin B12, but the clinical relevance of this effect is not clear. Loss of gastric acidity also may affect the bioavailability of such drugs as ketoconazole, ampicillin esters, and iron salts. Chronic use of PPIs has been reported to be associated with an increased risk of bone fracture and with increased susceptibility to certain infections (e.g., hospital-acquired pneumonia, community-acquired Clostridium difficile). Hypergastrinemia is more frequent and more severe with PPIs than with H2 receptor antagonists. This hypergastrinemia may predispose to rebound hypersecretion of gastric acid upon discontinuation of therapy and also may promote the growth of GI tumors.
Therapeutic Uses. Prescription PPIs are used to promote healing of gastric and duodenal ulcers and to treat gastroesophageal reflux disease (GERD), including erosive esophagitis, which is either complicated or unresponsive to treatment with H2 receptor antagonists. Over-the-counter omeprazole is approved for the self-treatment of heartburn. PPIs also are the mainstay in the treatment of pathological hypersecretory conditions, including the Zollinger-Ellison syndrome. Lansoprazole and esomeprazole are approved for treatment and prevention of recurrence of NSAID-associated gastric ulcers in patients who continue NSAID use. It is not clear if PPIs affect the susceptibility to NSAID-induced damage and bleeding in the small and large intestine. All PPIs are approved for reducing the risk of duodenal ulcer recurrence associated with H. pylori infections. Therapeutic applications of PPIs are further discussed later under “Specific Acid-Peptic Disorders and Therapeutic Strategies.”
H2 RECEPTOR ANTAGONISTS
The H2 receptor antagonists inhibit acid production by reversibly competing with histamine for binding to H2 receptors on the basolateral membrane of parietal cells.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


