Chapter 6
Pharmacology of cell excitation
6.1 Overview
Nerve cells, muscle cells and several other cell types are excitable, that is, they are activated by variations of their membrane potentials. Many of the macromolecules involved in regulating cell excitation are important drug targets.
6.1.1 Clinical applications of drugs that influence cell excitation
- blockade of nerve conduction for local anesthesia
- reduction of brain cell excitability in epilepsy
- stabilization of mood in the treatment of bipolar disorder
- reduction of smooth muscle cell tone in arteries to reduce blood pressure
- suppression of aberrant excitation in cardiac arrhythmia
Notes: This list is not complete, but it suffices to illustrate that the applications are important and diverse. In order to understand how such drugs work, we must first understand cell excitation itself.
6.1.2 The nature of cell excitation
- All cells have an electrical potential across the cytoplasmic membrane, such that the cell interior is electrically negative relative to the outside (~−70 mV)
- In non-excitable cells, this membrane potential is stable; in excitable cells, it forms the resting potential
- Cell excitation consists in transient reversals of the membrane potential, called action potentials, which spread rapidly across the entire cell membrane
- Action potentials are spontaneously generated by some cells and transmitted between cells through chemical or electrical synapses
Notes: By definition, a negative membrane potential means that the cell interior is negative relative to the extracellular space. The negative resting potential is maintained by potassium leak channels in the cytoplasmic membrane; we will consider below how this works.
6.1.3 Neurons and synapses
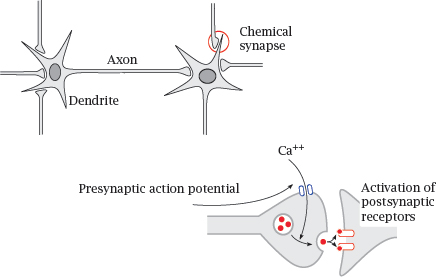
Notes: Many drugs that influence cell excitation act on nerve cells. Nerve cells, or neurons, communicate with one another through chemical synapses. A nerve cell receives input from other nerve cells upstream through synapses on its dendrite. When stimulated sufficiently by activity of these synapses, it will generate an action potential. The action potential travels down the dendrite toward the cell body, then down the axon. When it reaches a presynaptic terminal, the action potential causes an influx of Ca++, which in turn triggers the release of neurotransmitter into the synaptic cleft. Activation of postsynaptic receptors then transmits the signal to the next nerve cell.
A given neuron receives input from its afferent synapses, and it sends its output to its efferent synapses.
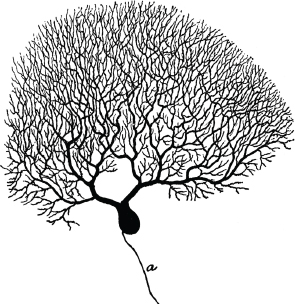
6.1.4 Some nerve cells have huge dendrites and axons
Notes: This picture (from [35]) shows a Purkinje cell from the cortex of the cerebellum. It has a large branched dendrite,1 through which it receives input from several thousand other nerve cells.
The axon of the Purkinje cell has far fewer branches than its dendrite (and they would all be below the lower edge of this picture). However, in some other types of nerve cells, the axon may be similarly strongly branched. For example, a single α-motoneuron may control the activity of several hundred skeletal muscle fibers; its axon must branch out and form synapses with each of these cells.
6.1.5 Other types of excitable cells
Notes: In addition to nerve cells, several other types of cells are also excitable and may be relevant as drug targets. Some of these cells receive their input from nerve cells. Adrenal gland cells release norepinephrine and epinephrine when stimulated by nerve cells of the autonomic nervous system, and skeletal muscle cells contract when stimulated by α-motoneurons.
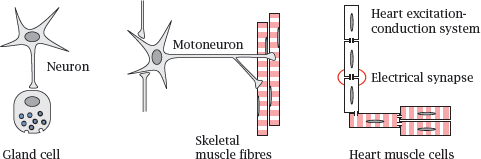
The heart, while also subject to modulatory input from the autonomic nervous system, can function entirely independently.2 It possesses two types of excitable cells. Those within the excitation-conduction system spontaneously produce the rhythm, which they then propagate to the regular “worker” muscle cells. Communication occurs through electrical synapses, in which ions can flow directly through cytoplasm bridges or gap junctions between cells.
6.2 The physical basis of cell excitation
Now comes the scary part—the one we all thought we had successfully dodged when choosing bioscience over real science.
6.2.1 The two driving forces that generate diffusion potentials across membranes
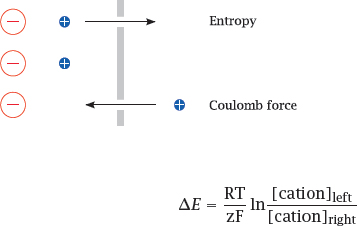
Notes: A diffusion potential arises if (1) the membrane is selectively permeable for some ion but not its counterion, and (2) there is a concentration gradient for the permeant ion.
Under these conditions, entropy will induce the permeant ion to move downhill its concentration gradient, until the ensuing charge imbalance creates an equally strong Coulomb counterforce. This counterforce is reached at the equilibrium potential, E0, which is determined by the Nernst equation.3
6.2.2 Equilibrium potentials for the major salt ions
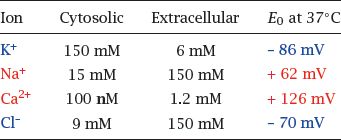
Notes: This table lists the major cations and anions that control the membrane potential and their intra- and extracellular concentrations. For each ion, the equilibrium potential was calculated using the Nernst equation.
As you can see, the equilibrium potentials are all different; those of K+ and Cl− are negative, while the ones for Na+ and Ca++ are positive. This raises the question: How do these different equilibrium potentials interact to shape the actual membrane potential?
Pure phospholipid membranes are essentially impermeable for salt ions; therefore, in cell membranes, ion permeabilities are determined by specific transport proteins, the ion channels. Ion channels that open and close and thereby cause ion permeabilities to change are at the heart of cell excitation.
6.2.3 Specific channels control ion permeabilities
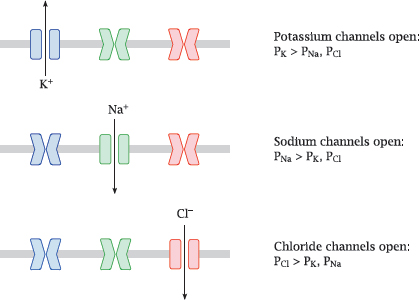
Notes: Some ion channels are continuously open, but most can open and close. Among those channels that open reversibly, ligand-gated channels open in response to binding of a chemical agonist, while voltage-gated channels respond to changes in the membrane potential. Some channels may respond to both stimuli. Yet others respond to changes in temperature or membrane distension; such channels are significant in sensing heat and pressure.
Many channels are very specific for individual ion species, as shown in this illustration; this applies to the major voltage-gated channels that control the action potential. Other channels have broader selectivity; for example, the nicotinic acetylcholine receptor (slide 6.10.1) is permeable for all of the major cations (Na+, K+ and Ca++).
In this slide, P represents the ion-specific membrane permeabilities that are explained in the next slide.
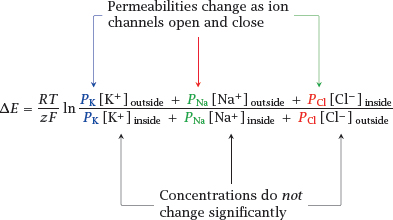
6.2.4 Diffusion potentials with multiple ions: the Goldman equation
Notes: The diffusion potential that prevails at a membrane that is permeable to several ion species is governed by the Goldman equation, shown here for a combination of Na+, K+ and Cl−. In addition to the concentration, it introduces a scaling parameter for each ion species, the permeability P, which weights each ion for its rate of diffusion across the membrane, relative to that of the other ions.
Transient reversals of the membrane potential are referred to as action potentials. In nerve and skeletal muscle cells, they last only 1-3 milliseconds, and the total number of ions that cross the membrane during an individual action potential is very low. With Na+, K+ and Cl−, it is too low to perceivably change the ion concentration on either side of the membrane.
The one important exception from this rule is calcium. Its intracellular concentration is so low (see slide 6.2.2) that the small number of ions flowing across open channels still causes a significant relative increase. This increase will cause activation of calmodulin and other calcium-binding proteins. Therefore, the opening of calcium channels constitutes both a physical and a biochemical signal.
6.3 Structure and function of voltage-gated cation channels
Voltage-gated cation channels are crucial in triggering, sustaining and propagating action potentials. The channels for K+, Na+ and Ca++ are homologous and functionally similar. We will look at the structure of voltage-gated K+ channels as an example.
6.3.1 Structure of a voltage-gated K+ channel
Notes: The picture on the left shows a side view. The gray rectangle delineates the membrane-embedded portion. A large part of the channel protein protrudes into the cytosol. The channel is composed of four identical subunits; one is shown in blue, another one in yellow, and two in white. The conformationally flexible N-terminal inactivation domain that is located in the cytosolic portion (see below) is missing from this structure.
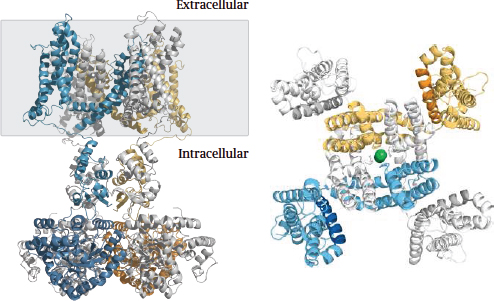
The panel on the right shows a view from the extracellular side onto the membrane-embedded domain of the channel. This domain consists of an inner layer, which contains the selectivity filter, and an outer layer that mediates voltage-dependent channel opening. In each of the four subunits, an arginine-rich helix that forms the voltage sensor is rendered in a darker shade. A potassium ion inside the central channel pore is shown as a green ball. Structure from [36].
6.3.2 Structure of the K+ selectivity filter
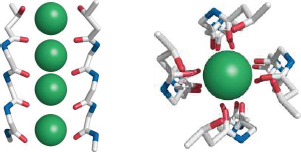
Notes: Side view (left) and top view (right) of the K+ selectivity filter that is in the center of the membrane-embedded portion. Backbone oxygen atoms tightly enclose the dehydrated K+ ions. Each of the four subunits contributes one backbone segment, but for clarity only two segments are shown in the side view.
Similar selectivity filters are also found in other types of K+ channels, for example the leak channels that maintain the resting potential, as well as the KATP channels (slide 6.6.1).
The selectivity filters found in sodium and calcium channels work according to the same principle, although they differ in diameters and architectural detail. In each case, the filter fits the ion like a glove. Ions that are too large are simply excluded; those that are too small are rejected because they cannot bind avidly to the filter, and therefore the energetic cost of their dehydration cannot be offset.
6.3.3 Voltage-gated sodium channels sustain and spread the action potential
Notes: On nerve and skeletal muscle cells, the action potential is carried and sustained mostly by voltage-gated Na+ (NaV) channels. As stated above, all voltage-gated channels have the same general structure as the KV channel, so we will now see how those structural features operate in practice.
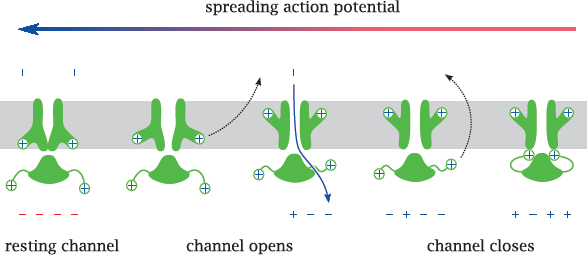
When the membrane potential in the vicinity of a sodium channel is reversed by a spreading action potential, the membrane-embedded voltage sensor moves outward, which decompresses the inner layer and opens the channel. This allows Na+ to enter the cell, enlarging the depolarized membrane area, causing more NaV to open and the action potential to propagate further.
After the channel has been open for a short time,4 a separate inactivation gate, which is flexibly attached to the cytosolic portion of the channel and also carries a positive net charge-and which, as stated, is missing from the KV channel structure shown above-responds to the same electric field reversal and moves to plug the channel. The channel is now inactivated, in spite of the continued depolarized state of the membrane. Before the channel becomes ready for opening again, both gates must revert to their initial conformations, which happens only after the membrane has been repolarized.5
6.3.4 Voltage-gated potassium channels extinguish the action potential
Notes: This slide illustrates the interplay of NaV and KV channels in shaping the action potential. Both channels open in response to depolarization. Because of the opposite orientations of the two concentration gradients, this causes Na+ influx but K+ efflux.
The NaV channels open and inactivate faster and therefore dominate the early phase of the action potential. The slower response of the KV channels dominates the later phase and causes a transient hyperpolarization of the membrane.
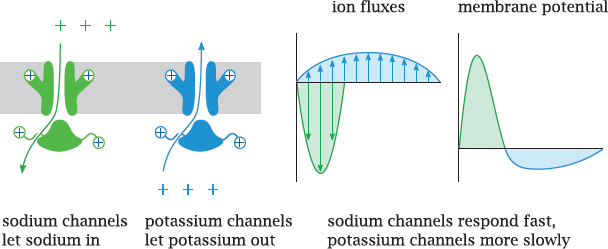
As long as the composition of the channel population remains the same, the shape and amplitude of each action potential will also be the same.6 Hence, information cannot be encoded in the properties of individual action potentials; instead, it is encoded in their frequency.
6.4 Measuring ion fluxes across single channels
One of the fascinations that ion channels hold for many researchers is the ease and detail with which the behavior of individual channel molecules can be studied. Here, we take a brief look at how this is done.
6.4.1 Planar lipid bilayers
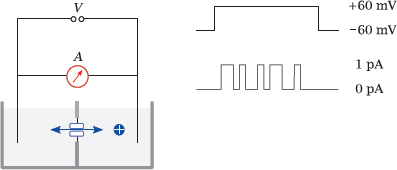
Notes: In this experimental setup, a single lipid bilayer is “painted” across a small aperture between two buffered reservoirs. Purified channel molecules, typically solubilized with some non-denaturing detergent, are introduced into one reservoir. Membrane proteins prepared in this way tend to spontaneously insert again into lipid bilayers when offered the opportunity; the right amount of protein sample that will cause just one channel molecule to find and insert into the membrane patch is determined by trial and error.
Electrodes are inserted into the buffer reservoirs on both sides of the bilayer. Voltage is applied, and the opening and closing of the channel can be observed, typically in the form of discreet jumps of constant magnitude that is proportional to the conductivity of the channel molecule.
The buffer solutions usually contain salts in high concentration (~1 M), since this will yield greater currents in response to channel opening. Even then, the currents are typically on the order of only 1 pA. Additional compounds can be introduced into either chamber to examine their effect on the channel from either end.
6.4.2 The patch clamp technique
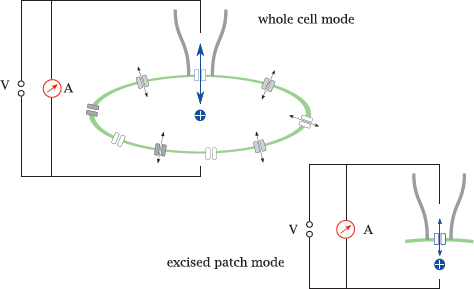
Notes: In the patch clamp technique, the channel is observed in its natural environment within the cell membrane; therefore, interactions with regulatory proteins or other effectors are preserved. Isolated observation of a single channel, or of a small number of channels, is accomplished by gently lowering a glass micro-pipette onto the cell surface, such that it forms a seal with the cell membrane. All current through the pipette must then flow through the membrane patch delineated by the pipette aperture, and through any channels this patch happens to contain.
In whole-cell mode, the current has to pass the membrane twice, once across the sealed patch, and once across the remainder of the cell surface. However, the latter contains very many channel molecules arranged in parallel, causing negligible ohmic resistance; therefore, the current is controlled almost exclusively by the channels in the patch. In excised patch mode, the pipette is withdrawn after seal formation; the cell membrane ruptures, with the pipette holding on to its adhering patch of membrane. This makes it possible to experimentally vary the buffer milieu on both the extracellular side and the intracellular side of the membrane.
Channels of different specificities will usually be present in the membrane patch, and in order to selectively observe one species, some others may need to be suppressed, through either addition of specific inhibitors, or elimination of their specific substrate ions from the buffer. For example, sodium and potassium channels may be suppressed by replacing Na+ and K+ with tetraethylammonium ions; this trick can be used to selectively detect calcium currents.
6.5 Drug effects on voltage-gated sodium (NaV) channels
Sodium channels play a crucial role in the propagation of action potentials, and thus they are a good target if inhibition of nerve conduction is desired, as it is in local anesthesia. In addition, inhibitors of sodium channels are also used in several other situations that require dampening of neural excitability.
6.5.1 Fast and slow channel block
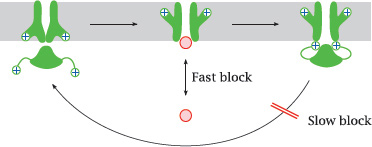
Notes: Channel-blocking drugs may cause either fast or slow blocks. A fast block occurs when a drug reversibly binds within the channel lumen and obstructs it.
A slow block is observed when a drug binds to the inactivated state of the channel and delays its reactivation. Both fast and slow blocks may be use-dependent, that is, the channel needs to be activated in order to allow the drugs to enter and block the channel molecule.
6.5.2 Diethylamine and phenol resemble parts of the lidocaine molecule
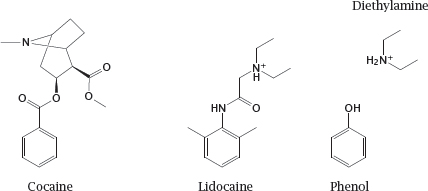
Notes: The drug lidocaine is used for local anesthesia and in the treatment of certain types of cardiac arrhythmias. It causes both slow and fast block at the NaV channel. The model compounds diethylamine and phenol resemble parts of the lidocaine molecule; as we will see in the next slide, they also mimic partial activities of it.
Cocaine also blocks sodium channels and can be used for local anesthesia, but it is no longer used in this application. This activity of cocaine is unrelated to its psychogenic effect that is caused in dopaminergic synapses (slide 6.14.3).
6.5.3 Effects of diethylamine and of phenol on NaV channel conductance
Notes: This slide shows a series of single channel recordings on NaV channels in planar lipid bilayers. The channels were biased toward the open state with batrachotoxin (see next slide). Diethylamine reversibly blocks the open state; the block is more pronounced if the membrane potential pulls the drug into the channel (as is the case with the +40 mV trace). Individual binding and dissociation events happen too fast for being resolved by the recording electronics, which therefore averages them out and reports an apparent decrease in the conductance. Such behavior constitutes a fast block.
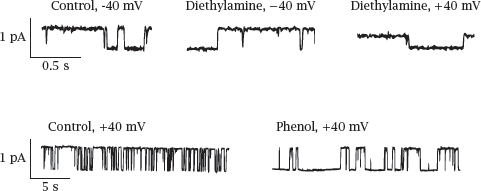
In contrast to diethylamine, phenol measurably extends the time intervals of channel inactivity but does not alter the observed conductivity of the open state. This represents a slow block. Thus, it appears that diethylamine and phenol respectively account for the dual fast and slow block that was reported previously for lidocaine. Figure adapted from [37].
6.5.4 Drugs and poisons that act on NaV channels
Notes: Batrachotoxin interacts with the channel from within the lipid bilayer and activates it. It has no therapeutic application but is useful in experimental studies on NaV channels.
Phenytoin is also used in epilepsy. In addition to blocking sodium channels, it also affects some types of potassium and calcium channels. This may give rise to cardiac arrhythmias but also contribute to its antiepileptic efficacy.
6.6 Pharmacology of potassium channels
In addition to voltage-gated potassium channels, there are several other families of potassium channels. K+ leak channels, which are always open, dominate the resting potential, and other channel types are regulated by various ligands, including Ca++ or G protein βγ-dimers (see slide 5.3).
A pharmacologically important K+ channel, which belongs to the Kir family,7 is associated with and controlled by another membrane protein, the sulfonylurea receptor. This receptor functions as an ATP sensor; when bound to ATP, it closes the channel. The complex comprising the channel and the receptor is referred to as the KATP channel.
6.6.1 KATP channels regulate the tone of smooth muscle cells
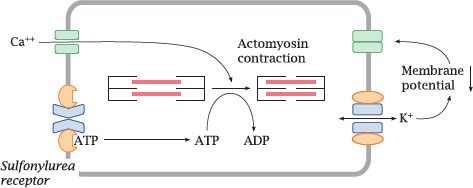
Notes: In vascular smooth muscle cells, the KATP channel serves to regulate the strength of contraction. Sustained contraction of the cell will deplete ATP. This will promote dissociation of ATP from the sulfonylurea receptor and open the connected Kir channel. The increase in K+ permeability will lower the membrane potential and inhibit the activation of CaV channels; this, in turn, will inhibit cell contraction. If a smooth muscle cell is exhausted and depleted of ATP, opening of the KATP channels gives it a way to “tune out” calcium signals and suspend contraction until it has caught its breath and replenished ATP.
6.6.2 KATP channels in pancreatic β cells regulate insulin secretion
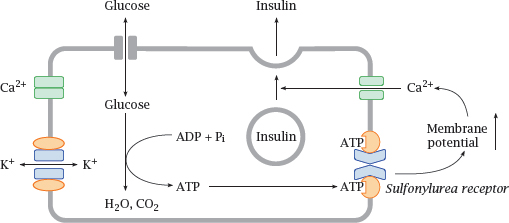
Notes: KATP channels also occur in the β cells of pancreatic islets, where they regulate the release of insulin depending on the blood glucose level. While most cells require insulin for the uptake of glucose, the islet cells themselves do not; therefore, the intracellular glucose levels will rise and fall with blood glucose.
When blood glucose is high, the islet cells will take up more of it and then degrade it. This produces ATP, which binds the sulfonylurea effector, closes Kir, increases the membrane potential and thereby promotes calcium-mediated insulin release. Pharmacological activation of the sulfonylurea receptor in order to increase insulin secretion is a useful strategy in type 2 diabetics, who, in contrast to type 1 diabetics, retain the ability to produce their own insulin.
Note that the intended effects on the sulfonylurea receptor differ between the two applications—in type 2 diabetics, we want to inhibit the receptor, whereas in hypertension we want to activate it. This leads to mutual side effects of antidiabetic and antihypertensive therapy. The two channel molecules are slightly different, which may be exploited by selective drugs to reduce this mutual interference.
6.6.3 Drugs that act on K+ channels
Notes: Tolbutamide is a sulfonylurea receptor agonist and closes the associated Kir channel. It promotes insulin secretion from pancreatic β cells and is used for oral therapy in type II diabetics. (In the structure of tolbutamide, the sulfonylurea moiety is highlighted.)
Diazoxide and minoxidil sulfate are sulfonylurea receptor antagonists. They open Kir channels on vascular smooth muscle cells, which induces relaxation and lowers the blood pressure. The experimental drug iptakalim reportedly opens Kir channels in the vasculature but closes those in pancreatic β cells.
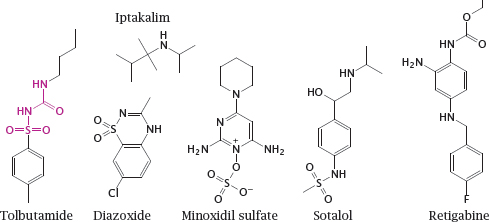
Sotalol inhibits KV channels, which delays repolarization and prolongs the action potential. It also blocks β-adrenergic receptors. The combination of these two effects can be useful in certain forms of cardiac arrhythmias. Retigabine activates neuronal KCNQ channels, which also belong to the KV family. It reduces neuronal excitability and is used to treat epilepsy.
6.7 Pharmacology of calcium channels
Calcium channels have a prominent role in controlling the rhythm and contraction of the heart, the activity of vascular smooth muscle, and the release of neurotransmitters from presynaptic terminals. All of these physiological effects have found pharmacological applications. We will first learn a bit more about the physiology of calcium channels and then look at some example drugs.
6.7.1 Voltage-gated channels and action potentials in the heart
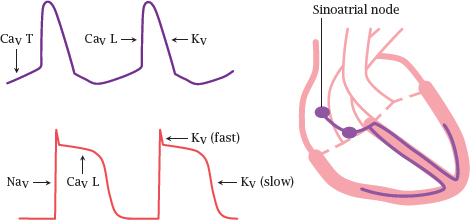
Notes: In the heart, we find two types of excitable cells: those in the excitation-conduction system generate action potentials spontaneously and periodically, whereas the “worker” muscle cells stay put until they are ordered otherwise, much like the muscle fibers in skeletal muscle.
In a healthy heart, action potentials are generated in the sino-atrial node, which is topmost center of the excitation-conduction system. From there, the action potentials travel through the remainder of the system and then spread to the worker cells.8
In the sinoatrial node, T type CaV channels open slowly and spontaneously at the resting potential, causing a gradual depolarization. Once this depolarization reaches the firing level of the faster and more abundant L type CaV channels, these open rapidly and trigger an action potential.
In the worker muscle cells, the action potential is initiated by NaV channels and sustained for the duration of the contraction through CaV channels.9 Repolarization is mediated by KV channels and L channel inactivation.
Drugs that affect T type CaV channels will preferentially act on the excitation-conduction system, whereas sodium channel blockers such as lidocaine will act mostly on the worker cells. Drugs that act on K+ channels or L type calcium channels will affect both cell types.
6.7.2 Two Ca++ channels control the contraction of striated muscle cells
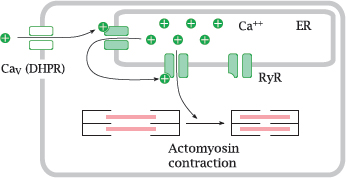
Notes: L-type CaV channels are also referred to as the dihydropyridine receptors (DHPRs); they control contraction both in the heart and in skeletal muscle cells.
When an action potential arrives and opens the channel, calcium flows in and opens the ryanodine receptor (RyR), a calcium-activated calcium channel that sits in the membrane of the endoplasmic reticulum. The large secondary wave of calcium released by the open RyR then triggers actomyosin contraction.
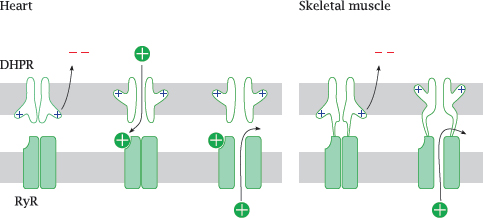
6.7.3 Entry of Ca++ through the DHPR is necessary in the heart, but not in skeletal muscle cells
Notes: There exists a surprising difference in the interaction of the two calcium channels between heart and skeletal muscle. In the heart, activation of the ryanodine receptor (RyR) requires actual influx of Ca++ through the dihydropyridine receptor (DHPR), which opens during an action potential.
In skeletal muscle, the two channels are hooked up directly to each other. Through this connection, the conformational change that occurs in the DHPR upon membrane depolarization is communicated to the RyR and causes it to open. Therefore, no actual influx of extracellular Ca++ is necessary—contraction can still occur even if the DHPR channel is plugged up, as long as it still performs its conformational pantomime of gate opening. Therefore, DHPR channel blockers will not interfere with contraction of skeletal muscle.