Application of Pharmacokinetics in Pharmacotherapeutics
By applying knowledge of pharmacokinetics to drug therapy, we can help maximize beneficial effects and minimize harm. Recall that the intensity of the response to a drug is directly related to the concentration of the drug at its site of action. To maximize beneficial effects, a drug must achieve concentrations that are high enough to elicit desired responses; to minimize harm, we must avoid concentrations that are too high. This balance is achieved by selecting the most appropriate route, dosage, and dosing schedule.
Passage of Drugs Across Membranes
All four phases of pharmacokinetics—absorption, distribution, metabolism, and excretion—involve drug movement. To move throughout the body, drugs must cross membranes. Drugs must cross membranes to enter the blood from their site of administration. When in the blood, drugs must cross membranes to leave the vascular system and reach their sites of action. In addition, drugs must cross membranes to undergo metabolism and excretion. Accordingly, the factors that determine the passage of drugs across biologic membranes have a profound influence on all aspects of pharmacokinetics.
Biologic membranes are composed of layers of individual cells. The cells composing most membranes are very close to one another—so close, in fact, that drugs must usually pass through cells, rather than between them, to cross the membrane. Hence the ability of a drug to cross a biologic membrane is determined primarily by its ability to pass through single cells.
Three Ways to Cross a Cell Membrane
The three most important ways by which drugs cross cell membranes are (1) passage through channels or pores, (2) passage with the aid of a transport system, and (3) direct penetration of the membrane. Of the three, direct penetration of the membrane is most common.
Channels and Pores
Very few drugs cross membranes through channels or pores. The channels in membranes are extremely small and are specific for certain molecules. Consequently, only the smallest of compounds, such as potassium and sodium, can pass through these channels, and then only if the channel is the right one.
Transport Systems
Transport systems are carriers that can move drugs from one side of the cell membrane to the other side. All transport systems are selective. Whether a transporter will carry a particular drug depends on the drug’s structure.
Transport systems are an important means of drug transit. For example, certain orally administered drugs could not be absorbed unless there were transport systems to move them across the membranes that separate the lumen of the intestine from the blood. A number of drugs could not reach intracellular sites of action without a transport system to move them across the cell membrane. One transporter, known as P-glycoprotein (PGP) or multidrug transporter protein, deserves special mention. PGP is a transmembrane protein that transports a wide variety of drugs out of cells.
Direct Penetration of the Membrane
For most drugs, movement throughout the body is dependent on the ability to penetrate membranes directly because (1) most drugs are too large to pass through channels or pores, and (2) most drugs lack transport systems to help them cross all of the membranes that separate them from their sites of action, metabolism, and excretion.
A general rule in chemistry states that “like dissolves like.” Membranes are composed primarily of lipids; therefore, to directly penetrate membranes, a drug must be lipid soluble (lipophilic).
Certain kinds of molecules are not lipid soluble and therefore cannot penetrate membranes. This group consists of polar molecules and ions.
Polar Molecules
Polar molecules are molecules that have no net charge; however, they have an uneven distribution of electrical charge. That is, positive and negative charges within the molecule tend to congregate separately from one another. Water is the classic example. As depicted in Fig. 4.1, the electrons (negative charges) in the water molecule spend more time in the vicinity of the oxygen atom than in the vicinity of the two hydrogen atoms. As a result, the area around the oxygen atom tends to be negatively charged, whereas the area around the hydrogen atoms tends to be positively charged. In accord with the “like dissolves like” rule, polar molecules will dissolve in polar solvents (such as water) but not in nonpolar solvents (such as lipids).
Ions
Ions are defined as molecules that have a net electrical charge (either positive or negative). Except for very small molecules, ions are unable to cross membranes; therefore, they must become nonionized in order to cross from one side to the other. Many drugs are either weak organic acids or weak organic bases, which can exist in charged and uncharged forms. Whether a weak acid or base carries an electrical charge is determined by the pH of the surrounding medium. Acids tend to ionize in basic (alkaline) media, whereas bases tend to ionize in acidic media. Therefore drugs that are weak acids are best absorbed in an acidic environment such as gastric acid because they remain in a nonionized form. When aspirin molecules pass from the stomach into the small intestine, where the environment is relatively alkaline, more of the molecules change to their ionized form. As a result, absorption of aspirin from the intestine is impeded.
Ion Trapping (pH Partitioning)
Because the ionization of drugs is pH dependent, when the pH of the fluid on one side of a membrane differs from the pH of the fluid on the other side, drug molecules tend to accumulate on the side where the pH most favors their ionization. Accordingly, because acidic drugs tend to ionize in basic media, and because basic drugs tend to ionize in acidic media, when there is a pH gradient between two sides of a membrane, the following occur:
The process whereby a drug accumulates on the side of a membrane where the pH most favors its ionization is referred to as ion trapping or pH partitioning.
Absorption
Absorption is defined as the movement of a drug from its site of administration into the systemic circulation. The rate of absorption determines how soon effects will begin. The amount of absorption helps determine how intense effects will be. Two other terms associated with absorption are chemical equivalence and bioavailability. Drug preparations are considered chemically equivalent if they contain the same amount of the identical chemical compound (drug). Preparations are considered equal in bioavailability if the drug they contain is absorbed at the same rate and to the same extent. It is possible for two formulations of the same drug to be chemically equivalent while differing in bioavailability. The concept of bioavailability is discussed further in Chapter 6.
Factors Affecting Drug Absorption
The rate at which a drug undergoes absorption is influenced by the physical and chemical properties of the drug and by physiologic and anatomic factors at the absorption site.
Rate of Dissolution
Before a drug can be absorbed, it must first dissolve. Hence, the rate of dissolution helps determine the rate of absorption. Drugs in formulations that allow rapid dissolution have a faster onset than drugs formulated for slow dissolution.
Surface Area
The surface area available for absorption is a major determinant of the rate of absorption. When the surface area is larger, absorption is faster. For this reason, absorption of orally administered drugs is usually greater from the small intestine rather than from the stomach. (Recall that the small intestine, because of its lining of microvilli, has an extremely large surface area, whereas the surface area of the stomach is relatively small.)
Blood Flow
Drugs are absorbed most rapidly from sites where blood flow is high because blood containing a newly absorbed drug will be replaced rapidly by drug-free blood, thereby maintaining a large gradient between the concentration of drug outside the blood and the concentration of drug in the blood. The greater the concentration gradient, the more rapid absorption will be.
Lipid Solubility
As a rule, highly lipid-soluble drugs are absorbed more rapidly than drugs whose lipid solubility is low. This occurs because lipid-soluble drugs can readily cross the membranes that separate them from the blood, whereas drugs of low lipid solubility cannot.
pH Partitioning
pH partitioning can influence drug absorption. Absorption will be enhanced when the difference between the pH of plasma and the pH at the site of administration is such that drug molecules will have a greater tendency to be ionized in the plasma.
Characteristics of Commonly Used Routes of Administration
For each of the major routes of administration—oral (PO), intravenous (IV), intramuscular (IM), and subcutaneous (subQ)—the pattern of drug absorption (i.e., the rate and extent of absorption) is unique. Consequently, the route by which a drug is administered significantly affects both the onset and the intensity of effects. The distinguishing characteristics of the four major routes are summarized in Table 4.1. Additional routes of administration (e.g., topical, transdermal, inhaled) each have unique characteristics that are addressed throughout the book as we discuss specific drugs that employ them.
TABLE 4.1
Properties of Major Routes of Drug Administration
| Route | Barriers to Absorption | Absorption Pattern | Advantages | Disadvantages |
| PARENTERAL | ||||
| Intravenous (IV) | None (absorption is bypassed) | Instantaneous | ||
| Intramuscular (IM) | Capillary wall (easy to pass) | |||
| Subcutaneous (subQ) | Same as IM | Same as IM | Same as IM | Same as IM |
| ENTERAL | ||||
| Oral (PO) | Epithelial lining of gastrointestinal tract; capillary wall | Slow and variable | ||
Distribution
Distribution is defined as the movement of drugs from the systemic circulation to the site of drug action. Drug distribution is determined by three major factors: blood flow to tissues, the ability of a drug to exit the vascular system, and, to a lesser extent, the ability of a drug to enter cells.
Blood Flow to Tissues
In the first phase of distribution, drugs are carried by the blood to the tissues and organs of the body. The rate at which drugs are delivered to a particular tissue is determined by blood flow to that tissue. Because most tissues are well perfused, regional blood flow is rarely a limiting factor in drug distribution.
There are two pathologic conditions—abscesses and tumors—in which low regional blood flow can affect drug therapy. An abscess has no internal blood vessels; therefore, because abscesses lack a blood supply, antibiotics cannot reach the bacteria within. Accordingly, if drug therapy is to be effective, the abscess must usually be surgically drained.
Solid tumors have a limited blood supply. Although blood flow to the outer regions of tumors is relatively high, blood flow becomes progressively lower toward the core. As a result, it may not be possible to achieve high drug levels deep inside tumors. Limited blood flow is a major reason that solid tumors are resistant to drug therapy.
Exiting the Vascular System
After a drug has been delivered to an organ or tissue by blood circulation, the next step is to exit the vasculature. Because most drugs do not produce their effects within the blood, the ability to leave the vascular system is an important determinant of drug actions. Drugs in the vascular system leave the blood at capillary beds.
Typical Capillary Beds
Most capillary beds offer no resistance to the departure of drugs because, in most tissues, drugs can leave the vasculature simply by passing through pores in the capillary wall. Because drugs pass between capillary cells rather than through them, movement into the interstitial space is not impeded. The exit of drugs from a typical capillary bed is depicted in Fig. 4.2.
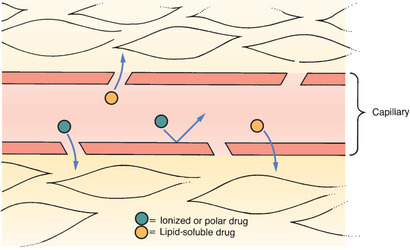
The Blood-Brain Barrier
The term blood-brain barrier (BBB) refers to the unique anatomy of capillaries in the central nervous system (CNS). As shown in Fig. 4.3, there are tight junctions between the cells that compose the walls of most capillaries in the CNS. These junctions are so tight that they prevent drug passage. Consequently, to leave the blood and reach sites of action within the brain, a drug must be able to pass through cells of the capillary wall. Only drugs that are lipid soluble or have a transport system can cross the BBB to a significant degree.
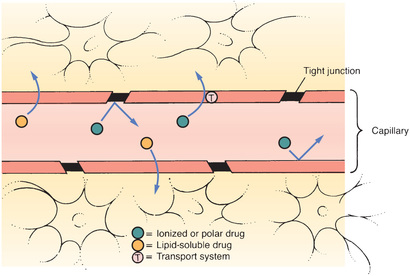
Recent evidence indicates that, in addition to tight junctions, the BBB has another protective component: PGP. As noted earlier, PGP is a transporter that pumps a variety of drugs out of cells. In capillaries of the CNS, PGP pumps drugs back into the blood and thereby limits their access to the brain.
The BBB is not fully developed at birth. As a result, newborns have heightened sensitivity to medicines that act on the brain. Likewise, neonates are especially vulnerable to CNS toxicity.
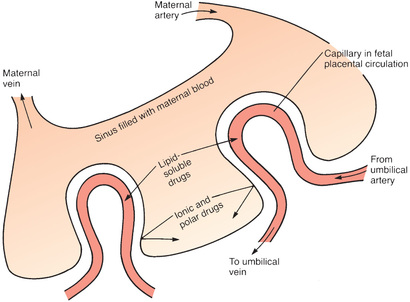
Placental Drug Transfer
The membranes of the placenta separate the maternal circulation from the fetal circulation (Fig. 4.4). However, the membranes of the placenta do NOT constitute an absolute barrier to the passage of drugs. The same factors that determine the movement of drugs across other membranes determine the movement of drugs across the placenta. Accordingly, lipid-soluble, nonionized compounds readily pass from the maternal bloodstream into the blood of the fetus. In contrast, compounds that are ionized, highly polar, or protein bound are largely excluded—as are drugs that are substrates for the PGP transporter that can pump a variety of drugs out of placental cells into the maternal blood.
Protein Binding
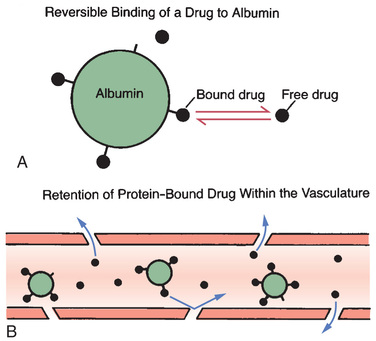
Drugs can form reversible bonds with various proteins in the body. Of all the proteins with which drugs can bind, plasma albumin is the most important. Like other proteins, albumin is a large molecule. Because of its size, albumin is too large to leave the bloodstream.
Fig. 4.5 depicts the binding of drug molecules to albumin. Note that the drug molecules are much smaller than albumin. As indicated by the two-way arrows, binding between albumin and drugs is reversible. Hence, drugs may be bound or unbound (free).
Even though a drug can bind albumin, only some molecules will be bound at any moment. The percentage of drug molecules that are bound is determined by the strength of the attraction between albumin and the drug. For example, the attraction between albumin and the anticoagulant warfarin is strong, causing nearly all (99%) of the warfarin molecules in plasma to be bound, leaving only 1% free. On the other hand, the attraction between the antibiotic gentamicin and albumin is relatively weak; less than 10% of the gentamicin molecules in plasma are bound, leaving more than 90% free.
An important consequence of protein binding is restriction of drug distribution. Because albumin is too large to leave the bloodstream, drug molecules that are bound to albumin cannot leave either (see Fig. 4.5B). As a result, bound molecules cannot reach their sites of action or undergo metabolism or excretion until the drug-protein bond is broken so that the drug is free to leave the circulation.
In addition to restricting drug distribution, protein binding can be a source of drug interactions. As suggested by Fig. 4.5A, each molecule of albumin has only a few sites to which drug molecules can bind. Because the number of binding sites is limited, drugs with the ability to bind albumin will compete with one another for those sites. As a result, one drug can displace another from albumin, causing the free concentration of the displaced drug to rise, thus increasing the intensity of drug responses. If plasma drug levels rise sufficiently, toxicity can result.
Entering Cells
Many drugs produce their effects by binding with receptors located on the external surface of the cell membrane; however, some drugs must enter cells to reach their sites of action, and practically all drugs must enter cells to undergo metabolism and excretion. The factors that determine the ability of a drug to cross cell membranes are the same factors that determine the passage of drugs across all other membranes, namely, lipid solubility, the presence of a transport system, or both.
Metabolism
Drug metabolism, also known as biotransformation, is defined as the enzymatic alteration of drug structure. Most drug metabolism takes place in the liver.
Hepatic Drug-Metabolizing Enzymes
Most drug metabolism that takes place in the liver is performed by the hepatic microsomal enzyme system, also known as the P450 system. The term P450 refers to cytochrome P450, a key component of this enzyme system.
It is important to appreciate that cytochrome P450 is not a single molecular entity, but rather a group of 12 closely related enzyme families. Three of the cytochrome P450 (CYP) families—designated CYP1, CYP2, and CYP3—metabolize drugs. The other nine families metabolize endogenous compounds (e.g., steroids, fatty acids). Each of the three P450 families that metabolize drugs is composed of multiple forms, each of which metabolizes only certain drugs. To identify the individual forms of cytochrome P450, designations such as CYP1A2, CYP2D6, and CYP3A4 are used to indicate specific members of the CYP1, CYP2, and CYP3 families, respectively.
Therapeutic Consequences of Drug Metabolism
Drug metabolism has six possible consequences of therapeutic significance:
Accelerated Renal Drug Excretion
The most important consequence of drug metabolism is promotion of renal drug excretion. The kidneys, which are the major organs of drug excretion, are unable to excrete drugs that are highly lipid soluble. Hence, by converting lipid-soluble drugs into more hydrophilic (water-soluble) forms, metabolic conversion can accelerate renal excretion of many agents.
Drug Inactivation
Drug metabolism can convert pharmacologically active compounds to inactive forms. This is the most common end result of drug metabolism.
Increased Therapeutic Action
Metabolism can increase the effectiveness of some drugs. For example, metabolism converts codeine into morphine. The analgesic activity of morphine is so much greater than that of codeine that formation of morphine may account for virtually all the pain relief that occurs after codeine administration.
Activation of Prodrugs
A prodrug is a compound that is pharmacologically inactive as administered and then undergoes conversion to its active form through metabolism. Prodrugs have several advantages; for example, a drug that cannot cross the BBB may be able to do so as a lipid-soluble prodrug that is converted to the active form in the CNS.
Increased or Decreased Toxicity
By converting drugs into inactive forms, metabolism can decrease toxicity. Conversely, metabolism can increase the potential for harm by converting relatively safe compounds into forms that are toxic. Increased toxicity is illustrated by the conversion of acetaminophen into a hepatotoxic metabolite. It is this product of metabolism, and not acetaminophen itself, that causes injury when acetaminophen is taken in overdose.
Special Considerations in Drug Metabolism
Several factors can influence the rate at which drugs are metabolized. These must be accounted for in drug therapy.
Age
The drug-metabolizing capacity of infants is limited. The liver does not develop its full capacity to metabolize drugs until about 1 year after birth. During the time before hepatic maturation, infants are especially sensitive to drugs, and care must be taken to avoid injury. Similarly, the ability of older adults to metabolize drugs is commonly decreased. Drug dosages may need to be reduced to prevent drug toxicity.
Induction and Inhibition of Drug-Metabolizing Enzymes
Drugs may be P450 substrates, P450 enzyme inducers, and P450 enzyme inhibitors. Drugs that are metabolized by P450 hepatic enzymes are substrates. Drugs that increase the rate of drug metabolism are inducers. Drugs that decrease the rate of drug metabolism are called inhibitors. Often a drug may have more than one property. For example, a drug may be both a substrate and an inducer.
Inducers act on the liver to stimulate enzyme synthesis. This process is known as induction. By increasing the rate of drug metabolism, the amount of active drug is decreased and plasma drug levels fall. If dosage adjustments are not made to accommodate for this, a drug may not achieve therapeutic levels.
Inhibitors act on the liver through a process known as inhibition. By slowing the rate of metabolism, inhibition can cause an increase in active drug accumulation. This can lead to an increase in adverse effects and toxicity.
First-Pass Effect
The term first-pass effect refers to the rapid hepatic inactivation of certain oral drugs. When drugs are absorbed from the gastrointestinal tract, they are carried directly to the liver through the hepatic portal vein before they enter the systemic circulation. If the capacity of the liver to metabolize a drug is extremely high, that drug can be completely inactivated on its first pass through the liver. As a result, no therapeutic effects can occur. To circumvent the first-pass effect, a drug that undergoes rapid hepatic metabolism is often administered parenterally. This permits the drug to temporarily bypass the liver, thereby allowing it to reach therapeutic levels in the systemic circulation before being metabolized.
Nutritional Status
Hepatic drug-metabolizing enzymes require a number of cofactors to function. In the malnourished patient, these cofactors may be deficient, causing drug metabolism to be compromised.
Competition Between Drugs
When two drugs are metabolized by the same metabolic pathway, they may compete with each other for metabolism and may thereby decrease the rate at which one or both agents are metabolized. If metabolism is depressed enough, a drug can accumulate to dangerous levels.
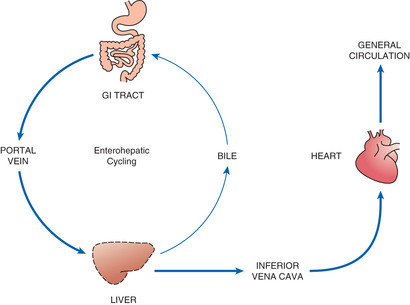
Enterohepatic Recirculation
As noted earlier and depicted in Fig. 4.6, enterohepatic recirculation is a repeating cycle in which a drug is transported from the liver into the duodenum (through the bile duct) and then back to the liver (through the portal blood). It is important to note, however, that only certain drugs are affected. Specifically, the process is limited to drugs that have undergone glucuronidation, a process that converts lipid-soluble drugs to water-soluble drugs by binding them to glucuronic acid. After glucuronidation, these drugs can enter the bile and then pass to the duodenum. In the intestine, some drugs can be hydrolyzed by intestinal beta-glucuronidase, an enzyme that breaks the bond between the original drug and the glucuronide moiety, thereby releasing the free drug. Because the free drug is more lipid soluble than the glucuronidated form, the free drug can undergo reabsorption across the intestinal wall, followed by transport back to the liver, where the cycle can start again. Because of enterohepatic recycling, drugs can remain in the body much longer than they otherwise would.
Excretion
Drug excretion is defined as the removal of drugs from the body. Drugs and their metabolites can exit the body in urine, bile, sweat, saliva, breast milk, and expired air. The most important organ for drug excretion is the kidney.
Renal Drug Excretion
The kidneys account for the majority of drug excretion. When the kidneys are healthy, they serve to limit the duration of action of many drugs. Conversely, if renal failure occurs, both the duration and intensity of drug responses may increase.
Steps in Renal Drug Excretion
Urinary excretion is the net result of three processes: (1) glomerular filtration, (2) passive tubular reabsorption, and (3) active tubular secretion.
Glomerular Filtration
Renal excretion begins at the glomerulus of the kidney tubule. As blood flows through the glomerular capillaries, fluids and small molecules—including drugs—are forced through the pores of the capillary wall. This process, called glomerular filtration, moves drugs from the blood into the tubular urine. Blood cells and large molecules (e.g., proteins) are too big to pass through the capillary pores and therefore do not undergo filtration. Because large molecules are not filtered, drugs bound to albumin remain in the blood.
Passive Tubular Reabsorption
As depicted in Fig. 4.7, the vessels that deliver blood to the glomerulus return to proximity with the renal tubule at a point distal to the glomerulus. At this distal site, drug concentrations in the blood are lower than drug concentrations in the tubule. This concentration gradient acts as a driving force to move drugs from the lumen of the tubule back into the blood. Because lipid-soluble drugs can readily cross the membranes that compose the tubular and vascular walls, drugs that are lipid soluble undergo passive reabsorption from the tubule back into the blood. In contrast, drugs that are not lipid soluble (ions and polar compounds) remain in the urine to be excreted.
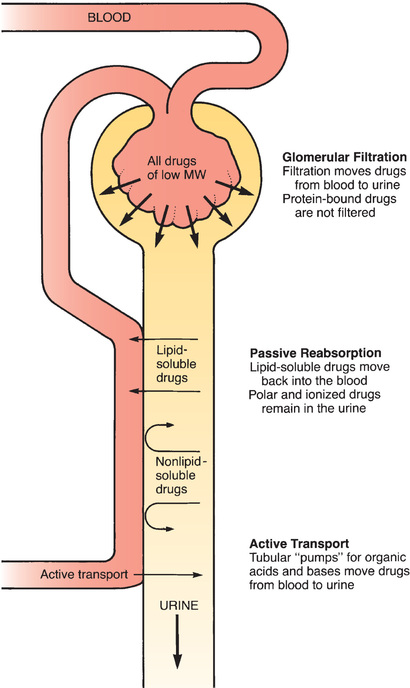
Active Tubular Secretion
There are active transport systems in the kidney tubules that pump drugs from the blood to the tubular urine. These pumps have a relatively high capacity and play a significant role in excreting certain compounds.
Factors That Modify Renal Drug Excretion
Renal drug excretion varies from patient to patient. Conditions such as chronic renal disease may cause profound alterations. Three other important factors to consider are pH-dependent ionization, competition for active tubular transport, and patient age.
pH-Dependent Ionization
The phenomenon of pH-dependent ionization can be used to accelerate renal excretion of drugs. Recall that passive tubular reabsorption is limited to lipid-soluble compounds. Because ions are not lipid soluble, drugs that are ionized at the pH of tubular urine will remain in the tubule and be excreted. Consequently, by manipulating urinary pH in such a way as to promote the ionization of a drug, we can decrease passive reabsorption back into the blood and can thereby hasten the drug’s elimination. This principle has been employed to promote the excretion of poisons as well as medications that have been taken in toxic doses.
Competition for Active Tubular Transport
Competition between drugs for active tubular transport can delay renal excretion, thereby prolonging effects. The active transport systems of the renal tubules can be envisioned as motor-driven revolving doors that carry drugs from the plasma into the renal tubules. These “revolving doors” can carry only a limited number of drug molecules per unit of time. Accordingly, if there are too many molecules present, some must wait their turn. Because of competition, if we administer two drugs at the same time, and if both drugs use the same transport system, excretion of each will be delayed by the presence of the other.
Age
The kidneys of newborns are not fully developed. Until their kidneys reach full capacity (a few months after birth), infants have a limited capacity to excrete drugs. This must be accounted for when medicating an infant.
In old age, renal function often declines. Older adults have smaller kidneys and fewer nephrons. The loss of nephrons results in decreased blood filtration. Additionally, vessel changes such as atherosclerosis reduce renal blood flow. As a result, renal excretion of drugs is decreased.
Nonrenal Routes of Drug Excretion
In most cases, excretion of drugs by nonrenal routes has minimal clinical significance. However, in certain situations, nonrenal excretion can have important therapeutic and toxicologic consequences.
Breast Milk
Some drugs taken by breast-feeding women can undergo excretion into milk. As a result, breastfeeding can expose the nursing infant to drugs. The factors that influence the appearance of drugs in breast milk are the same factors that determine the passage of drugs across membranes. Accordingly, lipid-soluble drugs have ready access to breast milk, whereas drugs that are polar, ionized, or protein bound cannot enter in significant amounts.
Other Nonrenal Routes of Excretion
The bile is an important route of excretion for certain drugs. Because bile is secreted into the small intestine, drugs that do not undergo enterohepatic recirculation leave the body in the feces.
The lungs are the major route by which volatile anesthetics are excreted. Alcohol is partially eliminated by this route.
Small amounts of drugs can appear in sweat and saliva. These routes have little therapeutic or toxicologic significance.
Time Course of Drug Responses
It is possible to regulate the time at which drug responses start, the time they are most intense, and the time they cease. Because the four pharmacokinetic processes—absorption, distribution, metabolism, and excretion—determine how much drug will be at its sites of action at any given time, these processes are the major determinants of the time course over which drug responses take place.
Plasma Drug Levels
In most cases, the time course of drug action bears a direct relationship to the concentration of a drug in the blood. Hence, before discussing the time course per se, we need to review several important concepts related to plasma drug levels.
Clinical Significance of Plasma Drug Levels
Providers frequently monitor plasma drug levels in efforts to regulate drug responses. When measurements indicate that drug levels are inappropriate, these levels can be adjusted up or down by changing dosage size, dosage timing, or both.
The practice of regulating plasma drug levels to control drug responses should seem a bit odd, given that (1) drug responses are related to drug concentrations at sites of action and (2) the site of action of most drugs is not in the blood. More often than not, it is a practical impossibility to measure drug concentrations at sites of action. Experience has shown that, for most drugs, there is a direct correlation between therapeutic and toxic responses and the amount of drug present in plasma. Therefore, although we cannot usually measure drug concentrations at sites of action, we can determine plasma drug concentrations that, in turn, are highly predictive of therapeutic and toxic responses. Accordingly, the dosing objective is commonly spoken of in terms of achieving a specific plasma level of a drug.
Two Plasma Drug Levels Defined
Two plasma drug levels are of special importance: (1) the minimum effective concentration, and (2) the toxic concentration. These levels are depicted in Fig. 4.8.
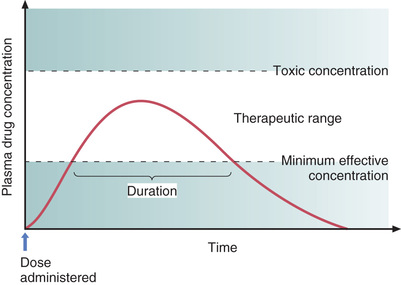
Minimum Effective Concentration
The minimum effective concentration (MEC) is defined as the plasma drug level below which therapeutic effects will not occur. Hence, to be of benefit, a drug must be present in concentrations at or above the MEC.
Toxic Concentration
Toxicity occurs when plasma drug levels climb too high. The plasma level at which toxic effects begin is termed the toxic concentration. Doses must be kept small enough so that the toxic concentration is not reached.
Therapeutic Range
As indicated in Fig. 4.8, there is a range of plasma drug levels, falling between the MEC and the toxic concentration, which is termed the therapeutic range. When plasma levels are within the therapeutic range, there is enough drug present to produce therapeutic responses but not so much that toxicity results. The objective of drug dosing is to maintain plasma drug levels within the therapeutic range.
The width of the therapeutic range is a major determinant of the ease with which a drug can be used safely. Drugs that have a narrow therapeutic range are difficult to administer safely. Conversely, drugs that have a wide therapeutic range can be administered safely with relative ease. The principle is the same as that of the therapeutic index discussed in Chapter 3. The therapeutic range is quantified, or measured, by the therapeutic index.
Understanding the concept of therapeutic range can facilitate patient care. Because drugs with a narrow therapeutic range are more dangerous than drugs with a wide therapeutic range, patients taking drugs with a narrow therapeutic range are the most likely to require intervention for drug-related complications. The provider who is aware of this fact can focus additional attention on monitoring these patients for signs and symptoms of toxicity.
Single-Dose Time Course
Fig. 4.8 shows how plasma drug levels change over time after a single dose of an oral medication. Drug levels rise as the medicine undergoes absorption. Drug levels then decline as metabolism and excretion eliminate the drug from the body.
Because responses cannot occur until plasma drug levels have reached the MEC, there is a latent period between drug administration and onset of effects. The extent of this delay is determined by the rate of absorption.
The duration of effects is determined largely by the combination of metabolism and excretion. As long as drug levels remain above the MEC, therapeutic responses will be maintained; when levels fall below the MEC, benefits will cease. Because metabolism and excretion are the processes most responsible for causing plasma drug levels to fall, these processes are the primary determinants of how long drug effects will persist.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


