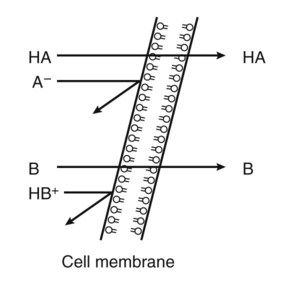
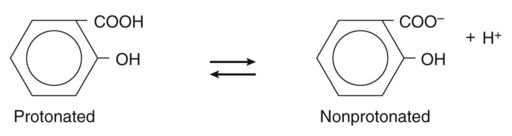
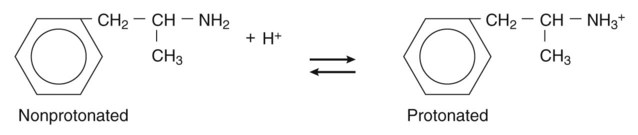
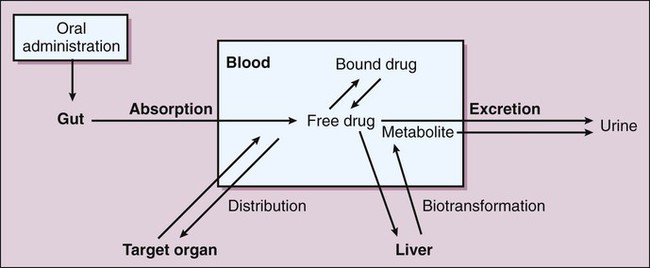
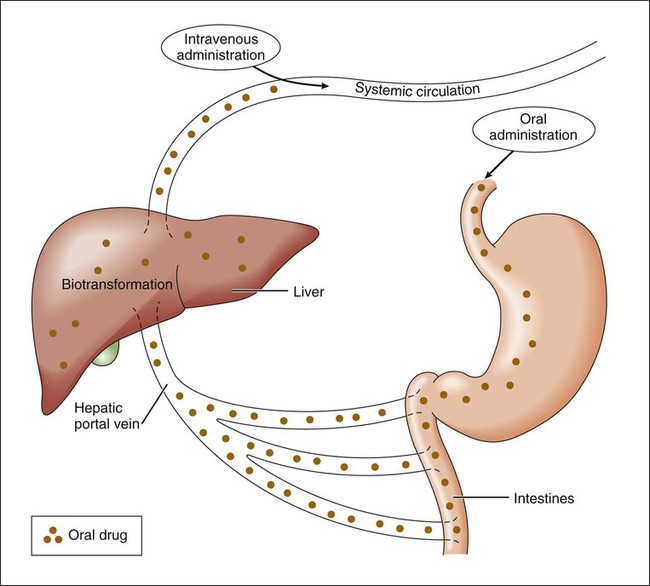
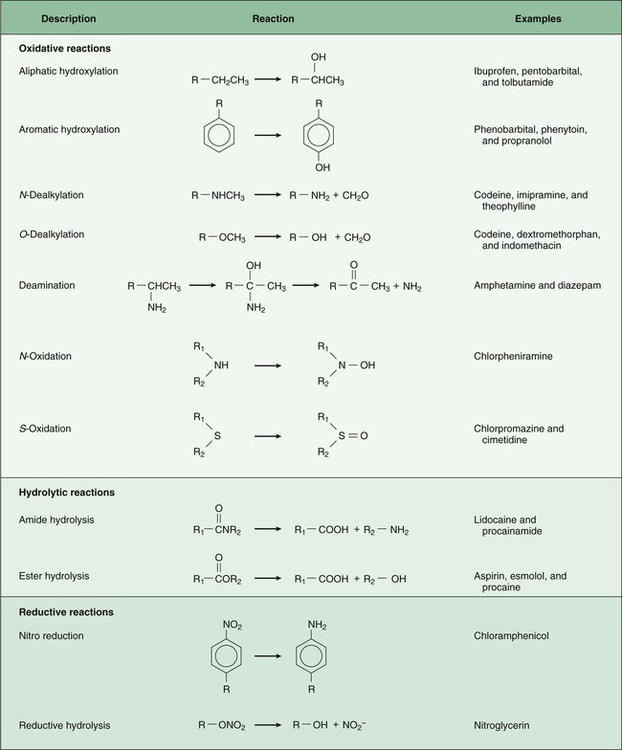
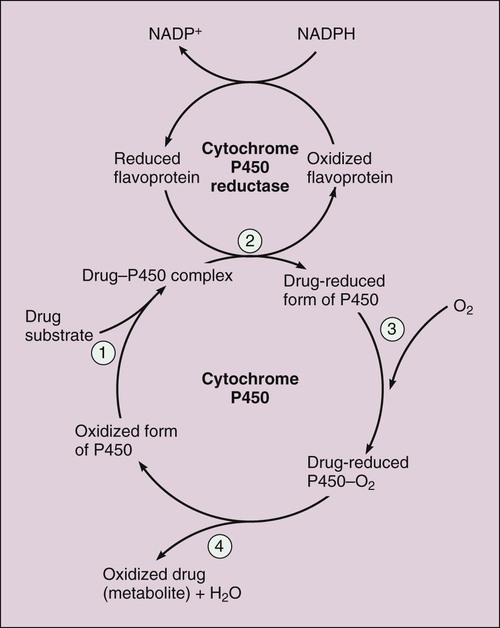
Pharmacokinetics is the study of drug disposition in the body and focuses on the changes in drug plasma concentration. For any given drug and dose, the plasma concentration of the drug will rise and fall according to the rates of three processes: absorption, distribution, and elimination. Absorption of a drug refers to the movement of drug into the bloodstream, with the rate dependent on the physical characteristics of the drug and its formulation. Distribution of a drug refers to the process of a drug leaving the bloodstream and going into the organs and tissues. Elimination of a drug from the blood relies on two processes: biotransformation (metabolism) of a drug to one or more metabolites, primarily in the liver, and the excretion of the parent drug or its metabolites, primarily by the kidneys. The relationship between these processes is shown in Figure 2-1. Many drugs are weak acids or bases that exist in both ionized and nonionized forms in the body. Only the nonionized form of these drugs is sufficiently soluble in membrane lipids to cross cell membranes (Box 2-1). The ratio of the two forms at a particular site influences the rate of absorption and is also a factor in distribution and elimination. Box 2-1 Effect of pH on the Absorption of a Weak Acid and a Weak Base Only the nonionized form of a drug can readily penetrate cell membranes. The protonated form of a weak acid is nonionized, whereas the protonated form of a weak base is ionized. The ratio of the protonated form to the nonprotonated form of these drugs can be calculated using the Henderson-Hasselbalch equation (see Box 2-1). The pKa is the negative log of the ionization constant, particular for each acidic or basic drug. At a pH equal to the pKa, equal amounts of the protonated and nonprotonated forms are present. If the pH is less than the pKa, the protonated form predominates. If the pH is greater than the pKa, the nonprotonated form predominates. Opposing the distribution of drugs to tissues are a number of ATP-driven drug efflux pumps, known as ABC transporters (ABC is an acronym for “ATP-binding cassette”). The most studied of these proteins, called permeability glycoprotein or P-glycoprotein (Pgp), is expressed on the luminal side of endothelial cells lining the intestines, brain capillaries, and a number of other tissues. Drug transport in the blood-to-lumen direction leads to a secretion of various drugs into the intestinal tract, thereby serving as a detoxifying mechanism. Pgp also serves to exclude drugs from the brain. The Pgp proteins exclude drugs from tissues throughout the body, including anticancer agents from tumors, leading to chemotherapeutic drug resistance. Inhibition of Pgp by amiodarone, erythromycin, propranolol, and other agents can increase tissue levels of these drugs and augment their pharmacologic effects (see Fig. 45-2). Plasma protein binding is saturable, and a drug can be displaced from binding sites by other drugs that have a high affinity for such sites. However, most drugs are not used at high enough plasma concentrations to occupy the vast number of plasma protein binding sites. There are a few agents that may cause drug interactions by competing for plasma protein binding sites, as highlighted in Chapter 4. Drugs that are absorbed from the gut reach the liver via the hepatic portal vein before entering the systemic circulation (Fig. 2-2). Many drugs, such as the antihypertensive agent felodipine (PLENDIL), are extensively converted to inactive metabolites during their first pass through the gut wall and liver, and have low bioavailability (see later) after oral administration. This phenomenon is called the first-pass effect. Drugs administered by the sublingual or rectal route undergo less first-pass metabolism and have a higher degree of bioavailability than do drugs administered by the oral route. Phase I biotransformation includes oxidative, hydrolytic, and reductive reactions (Fig. 2-3). The microsomal cytochrome P450 (CYP) monooxygenase system is a family of enzymes that catalyze the biotransformation of drugs with a wide range of chemical structures. The microsomal monooxygenase reaction requires the following: CYP (a hemoprotein); a flavoprotein that is reduced by nicotinamide adenine dinucleotide phosphate (NADPH), called NADPH CYP reductase; and membrane lipids in which the system is embedded. In the drug-oxidizing reaction, one atom of oxygen is used to form a hydroxylated metabolite of a drug, as shown in Figure 2-4, whereas the other atom of oxygen forms water when combined with electrons contributed by NADPH. The hydroxylated metabolite may be the end product of the reaction or serve as an intermediate that leads to the formation of another metabolite. Many drugs alter drug metabolism by inhibiting or inducing CYP enzymes, and drug interactions can occur when these drugs are administered concurrently with other drugs that are metabolized by CYP (see Chapter 4). Two examples of inducers of CYP are the barbiturate phenobarbital and the antitubercular drug rifampin. The inducers stimulate the transcription of genes encoding CYP enzymes, resulting in increased messenger RNA (mRNA) and protein synthesis. Drugs that induce CYP enzymes activate the binding of nuclear receptors to enhancer domains of CYP genes, increasing the rate of gene transcription. In phase II biotransformation, drug molecules undergo conjugation reactions with an endogenous substance such as acetate, glucuronate, sulfate, or glycine (Fig. 2-5). Conjugation enzymes, which are present in the liver and other tissues, join various drug molecules with one of these endogenous substances to form water-soluble metabolites that are more easily excreted. Except for microsomal glucuronosyltransferases, these enzymes are located in the cytoplasm. Most conjugated drug metabolites are pharmacologically inactive. Individuals exhibit slow or fast acetylation of some drugs because of genetically determined differences in N-acetyltransferase. Slow acetylators (SAs) were first identified by neuropathic effects of isoniazid, a drug to treat tuberculosis (see Chapter 41). These patients had higher plasma levels of isoniazid compared with other patients classified as rapid acetylators (RAs). The SA phenotype is autosomal recessive, although more than 20 allelic variants of the gene for N-acetyltransferase have been identified. In individuals with one wild-type enzyme and one faulty variant, an intermediate phenotype is observed. The distribution of these phenotypes varies from population to population. About 15% of Asians, 50% of Caucasians and Africans, and more than 80% of Mideast populations have the SA phenotype. Other drugs that may cause toxicity in the SA patient are sulfonamide antibiotics, the antidysrhythmic agent procainamide, and the antihypertensive agent hydralazine.
Pharmacokinetics
Overview
Drug Absorption
Effect of pH on Absorption of Weak Acids and Bases

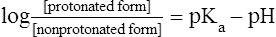

Drug Distribution
Factors Affecting Distribution
Organ Blood Flow
Plasma Protein Binding
Drug Biotransformation
First-Pass Biotransformation
Phases of Drug Biotransformation
Phase I Biotransformation
Oxidative Reactions
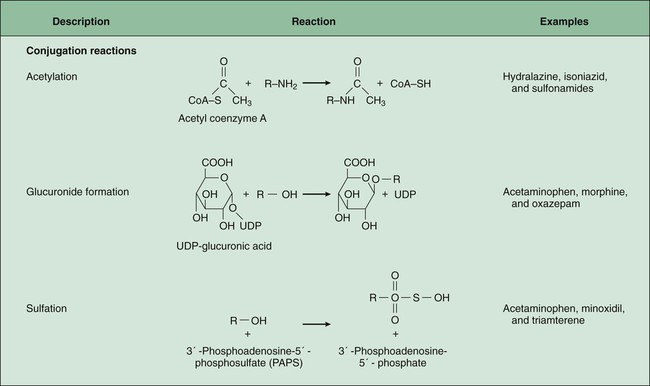
Phase II Biotransformation
Pharmacogenomics
Variations in Acetyltransferase Activity
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Pharmacokinetics
Only gold members can continue reading. Log In or Register to continue
