6
CHAPTER OUTLINE
Addiction medicine is based on the application of pharmacologic principles to drugs of abuse, ranging from prescription medications (e.g., narcotics, anxiolytics, stimulants) to Drug Enforcement Administration Class 1 compounds such as heroin, phencyclidine, and hallucinogenic mushrooms. Drugs of abuse are used to produce mood alterations; this requires drug penetration into the brain.
Pharmacologic principles in this chapter will focus on the pharmacokinetics of drug delivery to the brain; pharma-cogenomics, the genetic differences in metabolism, transport, and receptors that effect interindividual differences in drug disposition and response; and pharmacodynamic effects including tolerance and withdrawal.
BASIC PHARMACOLOGY CONCEPTS
Pharmacokinetics
Pharmacokinetics describes the time course of drug concentrations in blood and tissues (e.g., brain). Drug concentrations in blood and other sites are determined by absorption, distribution, metabolism, and elimination. The magnitude of a drug’s pharmacologic effect depends on the free (unbound) drug concentration at its site of action.
Absorption
Absorption is the process of drug movement from the site of drug delivery to the site of action. Psychoactive drugs can be taken orally (ethanol, amphetamines, barbiturates, opiates), intranasally (glue, solvents, amyl nitrate, cocaine, heroin), via smoking (nicotine, marijuana, freebase cocaine), intravenously (heroin, cocaine, methamphetamine), transdermally (fentanyl and nicotine patches), and subcutaneous injection. Figure 6-1 illustrates the differences in drug concentrations over time for the various routes of administration. The more rapidly a psychoactive drug is delivered to its site of action in the central nervous system, the greater is its mood-altering and reinforcing effects. The more rapidly achieved and higher the peak concentrations from intravenous and pulmonary (smoking) routes illustrate this point.
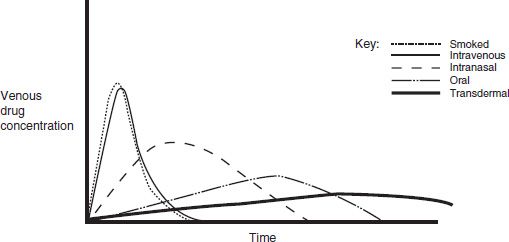
FIGURE 6-1 Venous drug concentrations after different routes of administration.
Bioavailability is defined as the fraction of unchanged drug that reaches the systemic circulation after administration by any route. The bioavailability factor (F) takes into account the portion of the administered dose that is able to enter the circulation unchanged. For intravenously administered drugs, F = 1.0 (100%). Bioavailability depends on a given drug’s site-specific membrane permeability, activity of drug transporters, and its first-pass metabolism. First-pass metabolism is the metabolism that occurs before a drug reaches the systemic circulation and occurs most extensively for lipid-soluble drugs such as morphine, methyl-phenidate, and desipramine and can significantly reduce bioavailability. Morphine, for example, requires nearly twice the dose when administered orally as compared to intravenously. First-pass metabolism is relatively unimportant for drugs administered through the intravenous, sublingual, intramuscular, subcutaneous, and transdermal routes, since drugs administered by these routes enter the general circulation directly.
For orally administered drugs, the rate of absorption is affected by (i) the pharmaceutical properties of the oral dosage form; (ii) the pH of gastric contents (drugs can be destroyed by extreme acid or basic conditions); (iii) gastric emptying time (faster gastric emptying time results in more rapid delivery to the small intestine, the site of absorption, except with dumping where the delivery to the small intestine is too fast to be optimally absorbed); (iv) intestinal transit time (for drugs absorbed in the small intestine, there is an increased rate of absorption with a faster intestinal transit time); (v) integrity of intestinal epithelium; and (vi) the presence of food (which decreases the interaction time between the drug and the intestinal villi) (1).
Efforts have been made to reformulate potentially addictive prescription drugs to reduce the rates of absorption in order to reduce the peak concentrations and reduce this reinforcing effect (2).
Smoked and inhaled drugs bypass the venous system and thus have the most rapid rate of delivery. Absorption of inhaled drug depends on the physical characteristics of the drug, including its volatility, particle size, and lipid solubility (3). Drugs that reach the alveoli of the lungs have rapid access to the bloodstream through closely applied capillary alveolar surfaces on the large pulmonary surface areas. Because a large portion of the cardiac output passes through the pulmonary circulation, the delivery of smoked and freebased drug to the brain is rapid.
Drugs must pass through biologic membranes to be absorbed. With passive diffusion, biologic membranes are more permeable to lipid-soluble and uncharged molecules. Some drugs have diminished absorption because of a reverse transporter associated with P-glycoprotein. This reverse transporter actively pumps drug out of the gut wall cells back into the gut lumen. When P-glycoprotein is inhibited, increased drug absorption results.
Some food and drug interactions alter first-pass metabolism and absorption from the intestinal wall. For example, components of grapefruit juice and other foods that either inhibit (e.g., grapefruit juice) or induce intestinal wall CYP3A4 or P-glycoprotein can lead to altered bioavailability of drugs that are substrates for this cytochrome (4,5). Also, the nonselective monoamine oxidase inhibitors (MAOIs) such as phenelzine and tranylcypromine—and, to a much lesser extent, the MAO B inhibitor selegiline and the reversible MAOI moclobemide—inhibit MAO A in the intestinal wall and liver. This inhibition diminishes the first-pass metabolism of tyramine, which is present in cheeses and various foods (6). When tyramine, an indirect-acting sympathomimetic amine, reaches the systemic circulation, it can produce increased release of norepinephrine from the sympathetic postganglionic neurons; this, in turn, can result in a severe pressor response and hypertensive crisis.
Hastening gastric emptying can help to achieve a more rapid drug effect without altering bioavailability. Gastric emptying can be hastened by taking a drug on an empty stomach with at least 200 mL of water and remaining in an upright position. Food, recumbency, heavy exercise, and drugs that slow gastric emptying (such as narcotics and anticholinergic drugs) can result in later and lower peak concentrations of the index drug.
Upon absorption, when drug concentrations are graphed against time, a peak drug concentration (Cmax) is reached at Tmax. The trough concentration is Cmin. The area under the concentration–time curve (AUC) is a measure of drug exposure that can be calculated and quantified.
Distribution
Once absorbed, a drug is distributed to the various organs and tissues of the body. Distribution is influenced by organ perfusion, organ size, binding of the drug within the blood and tissues, and the permeability of tissue membranes (7).
Most psychoactive drugs enter the brain because they are highly lipid soluble. The blood–brain barrier hinders the ability of non–lipid-soluble drugs to reach the brain tissue by diffusion (8). Unlike the fenestrated capillaries found throughout the body, which allow movement of molecules less than 25,000 Daltons, the endothelial cells lining brain capillaries have tight junctions and do not permit these small molecules to pass through. Without fenestrations, drugs must cross the two membranes of the endothelial cell by passive diffusion in order to enter the brain. The blood–brain barrier limits the admittance of many drugs to the brain and is found throughout the brain and spinal cord at all regions central to the arachnoid membrane, except for the floor of the hypothalamus and the area postrema, including the chemoreceptor trigger zone (where direct-acting chemicals can provoke vomiting).
For some compounds, however, specific active transport systems exist. These active transport systems enable glucose, amino acids, amines, purines, nucleosides, and organic acids to gain access to the brain (9). In contrast, P-glycoprotein is an efflux carrier present in the brain capillary endothelial cell, which bars the drug from translocating across the endothelial cell and actively exports the drug out of the brain (10).
When a drug distributes into all of the body compartments and tissues, it is said to distribute into an apparent volume of distribution (Vd). This volume has no direct physical equivalent because it describes the amount of serum, plasma, or blood that would be required to account for all the drug in the body. Vd can be thought of as the amount of drug in the body (D = dose) divided by the concentration of drug (C) in the plasma, or
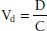
Drugs with a small Vd are confined primarily to the intra-vascular space of approximately 5 L. The drugs may be tightly bound to plasma proteins, or they may have a high molecular weight (large proteins, dextrans, and so forth). Drugs can have large Vd values up to 50,000 L if they are highly bound to tissue sites or are lipophilic.
Protein binding affects free (active) drug concentrations. Characteristics of binding proteins are capacity (amount of binding space) and affinity (tightness of binding interaction). Albumin is a high-capacity, low-affinity binding protein, whereas specific transport proteins such as transcortin are low capacity, high affinity. Acidic drugs commonly bind to albumin, the most abundant plasma protein; examples are barbiturates, benzodiazepines, and phenytoin. Basic drugs such as methadone bind to alpha1-acid glycoprotein, and others such as amitriptyline and nortriptyline bind to lipoproteins. Some binding sites are competitive, and a drug with a higher binding site affinity can displace a drug with a lower binding site affinity. Binding can also be stereospecific (specific for one stereoisomer of a compound). Drugs that are greater than 90% bound are considered highly protein bound, and reduced protein binding for these highly protein bound drugs can lead to large increases in drug effect.
The rate of blood flow delivered to specific organs and tissues affects drug distribution. Well-perfused tissues can receive large quantities of drug, provided that the drug can cross the membranes or other barriers present between the plasma and tissue. In contrast, poorly perfused tissues, such as fat, receive and release drug at a slow rate. This action explains why the concentration of drug in fat can be maintained long after the concentration in plasma has begun to decrease; anesthetics are examples of this phenomenon.
Clearance
Elimination refers to disappearance of the parent and/or active molecule from the bloodstream or body, which can occur by metabolism and/or excretion. Excretion is the process of removing a compound from the body without chemically changing that compound. Drugs can be excreted through the urine or feces, exhaled through the lungs, or secreted through sweat or salivary glands. The term clearance (Cl) represents the theoretical volume of blood or plasma that is completely cleared of drug in a given period of time. The factors that determine hepatic clearance are hepatic blood flow, the fraction of drug that is unbound, and the drug’s intrinsic clearance. If the intrinsic clearance of an unbound drug is very small, then this metabolic capacity (i.e., intrinsic clearance) of the liver, rather than hepatic blood flow, becomes the major determinant of hepatic clearance. In this case, functionality of hepatic enzymes determines drug clearance. If the intrinsic clearance of an unbound drug is very large, blood flow to the liver becomes rate limiting. Metabolic capacity determines drug clearance in most cases.
Most drugs display first-order elimination kinetics: the fraction or percentage of the total amount of drug present in the body removed at any one time is constant and independent of dose. Following administration of a drug with first-order kinetics, concentrations show an exponential decline of drug concentrations. The slope of this decay line is the elimination rate constant, kel, which is the percent of drug cleared per unit time (e.g., percent/hour). The half-life (t1/2) of a drug is the amount of time it takes for a drug concentration to decrease by half. One half-life represents a 50% change, and 2, 3, 4, and 5 half-lives represent 75%, 87.5%, 93.7%, and 96.8% changes, respectively. The time to reach steady state depends upon the duration of the half-life, whereas the amount of drug in the body at steady state will depend upon the frequency of drug administration and its dose. With drugs with dose-independent (first-order) disposition and elimination characteristics, five half-lives is a reasonable estimate of the time to reach steady state. For example, if the concentration at 2 hours postdose is 100 μg/mL, and the concentration at 4 hours postdose is 50 μg/mL, the t1/2 is 2 hours.
One means of calculating t1/2 is
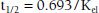
Using a more physiologic approach, t1/2 is directly related to the volume of distribution (Vd) and inversely related to the clearance (Cl). This relationship can be written as follows:
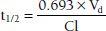
The constant 0.693 in this equation is derived from the natural logarithm of two [ln(2)]. Because drug elimination can be described by an exponential process, the time taken for a twofold decrease can be shown to be proportional to ln(2).
Although it is reasonable to assume that t1/2 and clearance are inversely related (clearance increases, so t1/2 decreases), effects of Vd on t1/2 do occur, which can offset the change in Cl (Vd decreases by the same proportion as clearance decreases, resulting in no change in t1/2).
In contrast, for drugs with zero-order elimination kinetics, the amount of drug removed (rather than the fraction of drug removed) at any one time is constant and dependent on dose. The maximal rate of metabolism and/or elimination is generally due to saturation of a key enzyme. This zero-order process is described by the Michaelis-Menten equation:
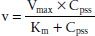
where v is the velocity of the reaction, Vmax is the maximum velocity of the reaction, Km is the concentration of the metabolic substrate when the velocity is ½Vmax, and Cpss is the steady-state concentration of drug. In the case of linear kinetics, Km is >>> Cpss, and v = Vmax × Cpss/(Km), or the v is proportional to the drug concentration: the higher the drug concentration, the faster the velocity. When Km <<< Cpss, v = Vmax, which is the case for drugs cleared by zero-order kinetics; the velocity is limited by Vmax. Because the half-life is inversely related to clearance, and clearance changes with drug concentration, the t1/2 is not constant. Therefore, half-life is not a useful descriptor for zero-order drugs. Drug dosing becomes difficult in these cases: a small increase in dose can cause a large increase in concentration, in contrast to drugs with first-order clearance where there is proportionality between dose and concentration. Aspirin, phenytoin, and ethanol are examples of drugs with zero-order elimination (11). See Figure 6-2 for a calculation of the decline in blood alcohol concentrations over time, demonstrating zero-order kinetics.
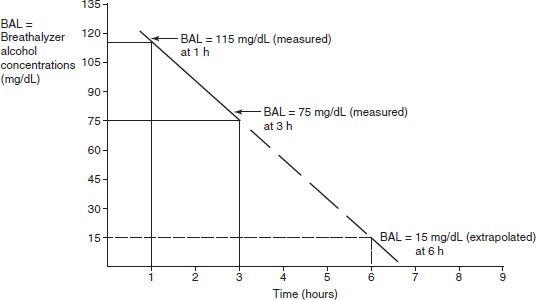
FIGURE 6-2 Extrapolating the decrease in blood alcohol concentration from two prior readings.
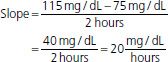
Expect at 4 hours BAL to be 55 mg/dL; 5 hours, BAL to be 35 mg/dL; 6 hours, BAL to be 15 mg/dL; and 7 hours, BAL to have already reached 0.00 mg/dL.
Drug metabolism is the process of chemical modification of drugs and other chemicals by the body, generally into less active and more hydrophilic compounds. These chemical modifications/reactions are generally performed by enzymatic systems, such as the cytochrome P450 enzyme system. Lipophilic drugs are generally transformed to more hydrophilic/polar products that are more readily eliminated by the kidney. Not all metabolites are inactive or nontoxic, and active metabolites need to be considered when assessing a drug’s total activity. In some cases, the administered drug is intentionally designed to be a pharmacologically inactive prodrug that is converted in vivo to a pharmacologically active molecule. Levodopa is one example, which (after crossing the blood–brain barrier) is converted in the basal ganglia to dopamine.
Drugs can be metabolized by Phase I and/or Phase II reactions. Phase I reactions are nonsynthetic reactions in which the drug is chemically altered and oxidized, for example, demethylated. Examples of Phase I reactions include the oxidation of phenobarbital, amphetamine, meperidine, and codeine by microsomal enzymes. Phase II reactions are synthetic reactions in which the drug is conjugated with another moiety, such as glucuronide or sulfate. Examples of synthetic reactions include the glucuronidation of morphine and meprobamate and acetylation of clonazepam and mescaline, which produce a compounds more polar than the parent drugs in order to facilitate elimination. Enzymes are responsible for most drug metabolism. The cytochrome P450 enzymes are most commonly involved and exist in the gut, liver, and brain. The gut and liver enzymes are the best studied. Oxidations can take place by cytochrome P450-dependent and cytochrome P450–independent mechanisms. Cytochrome P450–dependent oxidations include aromatic (phenytoin, amphetamine) and aliphatic (pento-barbital, meprobamate) hydroxylations, epoxidation, and oxidative dealkylation (morphine, caffeine, codeine), deam-ination (amphetamine), desulfurization (thiopental), and dechlorination. Cytochrome P450–independent oxidations include dehydrogenations (ethanol); azo, nitro, and carbonyl reductions (methadone and naloxone); and ester and amide hydrolysis (12).
More than 50 individual CYPs (pronounced “sip” for singular and “sips” for plural) have been identified in humans. As a family of enzymes, CYPs are involved not only in the metabolism of dietary and environmental compounds and medications but in the degradation of bile acids from cholesterol, the metabolism of retinoic and fatty acids including prostaglandins and eicosanoids, and the synthesis of steroids. A large number of specialized CYPs with specific substrate preferences are involved in these latter endogenous functions, in contrast to the relatively few numbers of CYPs that metabolize xenobiotics that have wider ranging and overlapping capabilities. CYPs that metabolize medications not only have a tremendous capacity to oxidize a large number of structurally diverse compounds but can metabolize a single compound at different positions on that molecule. Drug-metabolizing CYPs’ large and fluid substrate–binding sites contribute to their slow catalytic rates. In part, this explains why the half-lives of drugs are much longer than the half-lives of endogenous compounds.
CYPs are named with the root CYP followed by a number designating the family, a letter denoting the subfamily, and another number designating the CYP form. Thus, CYP2B6 is family 2, subfamily B, and gene number 6. Twelve CYPs (1A1, 1A2, 1B1, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 3A4, and 3A5) are known to metabolize xenobiotics in humans, and CYP3A4 alone is responsible for metabolizing more than 50% of clinically prescribed drugs.
Pharmacogenomics
Pharmacogenomics is the study of the relationship between genetic variations and drug disposition and response. The discipline of pharmacogenetics aims to elucidate cytochrome and other drug-metabolizing enzyme polymorphisms (different enzyme genetic subtypes), the degrees of expression of these polymorphisms, and the functional significance of such expression. Understanding these polymorphisms can help to explain individual differences in drug response.
Genetic variability in drug-metabolizing enzymes can affect drug bioavailability and clearance (13–16). Single nucleotide polymorphisms (SNPs) may alter CYP activity. CYP2D6, for example, which metabolizes codeine to morphine, is the best studied of the drug metabolic enzymes; 48 mutations and 50 alleles have been identified. Genotype and enzyme activity are linked to ethnicity, which varies from no gene/no enzyme activity (6% of whites) to two copies of a fully active gene (33% of Ethiopians). Individuals can be genotyped for 2D6 enzyme function (with classification as poor metabolizers [PMs], intermediate metabolizers [IMs], extensive metabolizers [EMs], and ultrarapid metabolizers [UMs]) (17). Those with PM genotypes generally do not receive adequate analgesia due to the inability to metabo-lize codeine to the active morphine. UMs, on the other hand, metabolize codeine significantly more rapidly and extensively than others, producing rare but life-threatening morphine intoxication. Breast-feeding infants of mothers who are UMs have received morphine overdoses from their mothers who are prescribed codeine for postpartum pain relief (18,19).
CYP2B6 has been less well studied. However, important genotypic influences on enzyme function have been identified, and examples of the importance of the variation in enzyme function on substrate metabolism follow. Several alleles have now been characterized as to enzyme amounts and activities, including CYP2B6 *1 (pronounced sip 2B6 star 1) (wild type), *2, *4, *5, *6, *9, and *18 (20). The *4 allele codes for an enzyme of higher function than the wild type. This allele has been associated with the toxicity of bupropion due to increased rates of conversion of bupro-pion to the active and longer-lasting metabolite, hydroxy-bupropion. Individuals with those genotypes were noted to have less enzyme present and lower enzyme activity as well. The effect of 2B6 genotype on bupropion-facilitated smoking cessation was reported by Zhu et al. (21). Their analysis determined that the hydroxylated metabolite contributed to the pharmacodynamic effect of smoking cessation and that in PMs an upward dosage adjustment could improve the rate of smoking cessation.
CYP3A4 has also had numerous SNPs identified. It appears that prediction of the activity of CYP3A4 is quite complex and not directly related to genotype (13). Several opioids including methadone and buprenorphine are metabolized by CYP3A4. Thus, the role of genetic polymorphisms in metabolism of CYP3A4 substrates is unclear at this time and an area of active research.
Prior to the more routine availability and use of genotyping, phenotyping of CYPs using marker compounds has been, and continues to be, a useful tool. A “cocktail” approach has been used, where subjects are administered microdoses of various compounds, each of which are metabolized by one specific enzyme and would reflect the enzymatic activity for clinically used medications. There are many “cocktail” recipes, one of which is the six-probe cocktail consisting of caffeine (1A2), flurbiprofen (2C9), mephenytoin (2C19), debrisoquine (2D6), chlorzoxazone (2E1), and dapsone (NAT2), where NAT2 is N-acetyltransferase 2 (22,23). A study of a pharmacokinetic interaction with antiretrovirals used midazolam as a marker of CYP3A4, fexof-enadine for P-glycoprotein, and pravastatin for OATP1B1 (organic anion transporter protein 1B1) (24). Many drugs, foods, and environmental chemicals can induce and/or inhibit the activity of the cytochromes, speeding up or slowing down their own metabolism (4,5). Each cytochrome isozyme responds differently to specific exogenous chemicals. For example, CYP1A1 is induced by polycyclic aromatic hydrocarbons, including benzo[α]pyrene contained in cigarette smoke.
Drug interactions at the level of the cytochromes and other metabolizing systems are often clinically significant. Methadone is metabolized primarily by CYP3A4, with contributions from CYP2B6, 2C19, 3A4, and, to a lesser extent, CYP2D6 (5,25). Inhibitors of CYP3A4, including erythro-mycin, diltiazem, ketoconazole, and saquinavir, slow the metabolism of methadone and increase methadone levels. Inducers of CYP3A4 such as carbamazepine, phenobarbital, efavirenz, and St. John’s wort speed the metabolism of methadone and decrease methadone levels. Awareness of potential interactions, clinical observation, and tailoring medication regimens and dosages are needed to optimize therapy and minimize potential toxicities.
Genetically defined differences in drug metabolism may influence risk of addiction, with relative protection for persons who experience adverse drug reactions at lower drug doses. Both the ADH1B2-His47Arg allele of alcohol dehydrogenase 1B and the ALDH-Glu487Lys allele of aldehyde dehydrogenase 2 alone or together can lead to flushing, nausea, and headache, owing to the accumulation of acetaldehyde when alcohol is consumed. Each of these alleles leads to a reduction in the risk of alcoholism, with an additive protective effect when the same person carries both alleles. Persons of South Asian descent are likely to carry both alleles, whereas those with Jewish ancestry often carry the Arg47 allele. Heterozygous carriers of ALDH2 Lys487 have low levels of ALDH2 enzyme activity, whereas ALDH2 Lys487/Lys487 homozygotes are nearly completely protected from alcoholism (26). Tyndale et al. (27) were among the first to postulate the existence of “pharmacogenetic protection factors.” They showed that a sample of whites who inherited two nonfunctional alleles for CYP2D6 were less likely to become dependent on oral opiates (estimated odds ratio >7). Slow nicotine metabolism by CYP2D6 appears to have a protective effect against nicotine addiction (28).
Although drug metabolism occurs largely in the liver, most other tissues and organs, including the lungs, gastrointestinal tract, skin, and kidneys, carry out varying degrees of drug metabolism. Understanding brain metabolism can be especially important in understanding the activity of psychoactive drugs (29). Many P450 cytochromes have been shown to catalyze the metabolism of neuros-teroids as well as psychoactive drugs such as neuroleptics and antidepressants. Alcohol produces a three- to fivefold increase in the level of brain P450 and induces CYP2C, CYP2E1, and CYP4A (30). Brain CYP2D6 can demethylate 3,4-methylenedioxy-N-methylamphetamine (MDMA or “ecstasy”) forming a harmful metabolite, N-methyl-a-methyldopamine. Brain CYP2D6 can O-demethylate para-methoxyamphetamine, a synthetic psychostimulant and hallucinogen, into 4-hydroxyamphetamine, which is also toxic (31). CYP2B6 metabolizes cocaine, phencyclidine, and some amphetamines. Both CYP2B6 and CYP2D6 are inducible by nicotine. P450s in other brain areas are induced by different factors (e.g., levels of CYP2C and CYP4A influence the activity of neurotransmitters such as dopamine, whichuse fatty acid metabolites as intracellular mediators).
Novel brain CYPs, such as 5α-androstane-3β, 17β-diol hydroxylase, CYP7B, and CYP2D4, continue to be discovered (32,33). Because the level of CYPs in the brain is approximately 0.5% to 2% of that in the liver and because brain CYP isoenzymes are of different types than those found in the liver, brain CYPs appear to be locally active but contribute little to overall pharmacokinetics of drugs in the body. The regulation of cytochrome P450 isozyme expression in the brain and elsewhere is being studied.
New areas of research include studies demonstrating novel brain transporter pathways (9,34). Epigenetic factors (35) and use of genome-wide association studies (36) may also yield new insights.
Pharmacodynamics
Pharmacodynamics is the study of the dose–response phenomena, which are the biochemical and physiologic effects of drugs on the body, and the body’s homeostatic response. Most drugs act on specific endogenous targets, or receptors, to modulate the rate and extent of the body’s endogenous functions. Receptors and their associated effector and transducer proteins coordinate signals from multiple ligands with the metabolic activities of the cell to act as integrators of this information.
Potency, Efficacy, and Dose–Response
When drug dose is plotted on a logarithmic scale, a sigmoi-dal curve often results, permitting mathematical manipulation of the results. The maximal efficacy of a drug occurs at Emax, its maximal effect. Efficacy is the extent of functional change imparted to a receptor. Efficacy is determined mainly by the nature of the receptor and its associated effector system. In contrast, potency is primarily determined by the affinity of the receptor for the drug. Potency denotes the amount of drug needed to produce a given effect. The concentration of the drug needed to produce 50% maximal effect occurs at EC50; the more potent the drug, the smaller the dose required to achieve maximal effect. In general, low potency is important only if the drug needs to be administered in undesirably large amounts. Because drug doses are readily adjusted, it is the maximal efficacy that is more often clinically relevant.
A similar sigmoidal curve is attained when the percentage of receptors that bind a drug is plotted against log drug concentration (Fig. 6-3). Here, the concentration at which 50% of the receptors are bound is denoted as Kd, and the maximum number of receptors bound is termed Bmax. Both “dose–response” and “dose–receptor-bound” graphs have a linear or nearly linear middle segment, indicating a first-order process. As the concentration of drug increases, a constant proportion of the drug binds to the receptor, causing a proportionate drug effect.
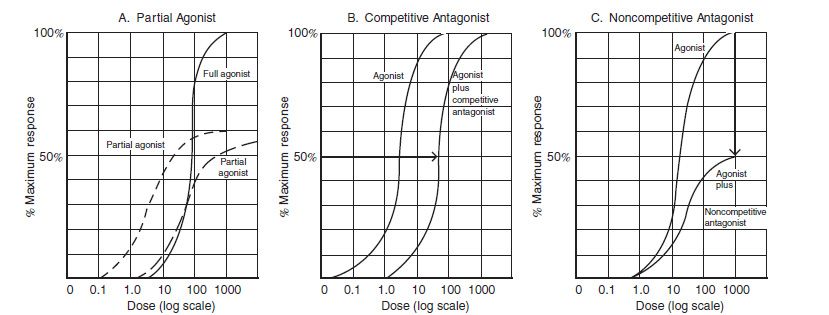
FIGURE 6-3 Full agonist, partial agonist, and competitive and noncompetitive antagonists. A: No matter how much the dose is increased, a partial agonist always will have a lower maximal efficiency (Emax). A partial agonist may be more potent, less potent, or equally potent as the agonist. In this example, both partial agonists decrease Emax. However, one partial agonist is more potent, and the other partial agonist is equi-potent, when compared to the full agonist. B: If there are no spare receptors, competitive agonists increase the EC50 but do not alter the other Emax. C: Noncompetitive antagonists decrease the Emax. (Modified from Katzung BG, Masters SB. Pharmacodynamics. In: Katzung and trevor’s pharmacology examination and board review. New York, NY: Lange Medical Books/McGraw-Hill, 2002:13.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


