Pharmacogenomics in Oncology
LEARNING OBJECTIVES
INTRODUCTION
Most types of cancer show considerable variability in their response to chemotherapy. In addition, anti-cancer therapies for treatment of a particular type of cancer, although significantly different in their mechanism of action, show only a marginal difference in outcome when compared with one another. This variability is thought to be due to inter-tumor and intra-tumor heterogeneity and host-specific factors and may reflect the lack of knowledge on how the molecular abnormalities in cancer cells affect responsiveness to anti-cancer therapies. In this aspect, pharmacogenomics can play an important role in predicting efficacy and toxicities of anti-cancer drugs.
Currently, there are over 100 drugs approved for treating cancer,1 and over 800 more in clinical development.2 Compared with other non-neoplastic diseases, selection of an anti-cancer treatment regimen is more critical since the mortality in patients increases significantly with the progression of disease.3 Due to a large number of therapeutic options, low response rates, high incidence of de novo and acquired resistance to therapies, and severe toxicities associated with anti-cancer agents, pharmacogenomic applications to develop predictive markers for drug response or toxicity are essential in oncology therapeutics.
Traditionally, most cytotoxic chemotherapy dosing is calculated with the use of weight, body surface area, or area under the curve. However, patients with inherited deficiencies in enzymes responsible for drug metabolism and disposition can have severe toxicities at these traditional doses. Patients who have a deficiency in thiopurine S-methyltransferase (TPMT) can have greatly elevated concentrations of active drug metabolites and are at risk for life-threatening, drug-induced myelosuppression. On the other hand, patients with increased enzymatic activity may be at risk for treatment failure resulting in cancer progression. Other examples of enzyme deficiencies and the drugs they metabolize include uridine diphosphate glucuronosyltransferase 1A1 (UGT1A1) and irinotecan, cytochrome P450 2D6 (CYP2D6) and tamoxifen, and dihydropyrimidine dehydrogenase (DPD) and 5-fluorouracil (5-FU).
In addition to pharmacogenomics of drug-metabolizing enzymes, there are mutations in drug targets that influence treatment outcome by conferring either sensitivity or resistance to therapy. One classical example is trastuzumab, a monoclonal antibody targeting human epidermal growth factor receptor 2 (ERBB2/HER2), which is overexpressed in some of the breast tumors. Trastuzumab resistance is seen in tumors that express p95, a truncated form of HER2, with no extracellular domain containing the trastuzumab binding site. In these cases, it is suggested that lapatinib may be a more effective treatment choice. Another example is breakpoint cluster region–v-abl Abelson murine leukemia viral oncogene homolog 1 (BCR-ABL)– targeted therapy in the treatment of chronic myeloid leukemia (CML). Treatment resistance to all the currently available BCR-ABL inhibitors (imatinib, dasatinib, and nilotinib) is seen when there is a T315I mutation.
Pharmacogenomic markers that predict response to treatment will allow clinicians to individualize treatment regimens based on patients’ genetic makeup. In the last decade there have been many advances and a variety of tests have been developed to see if a patient will respond to specific cancer therapies or will have a higher risk of toxicities. Examples include the K-RAS mutation test, where expression of the wild-type protein is associated with benefit from treatment with cetuximab, and the epidermal growth factor receptor (EGFR) mutation test, where gefitinib is only effective in those with activating mutations in the EGFR gene. In this chapter we provide a brief overview of cancer, present pertinent clinical cases demonstrating the importance of pharmacogenomics in oncology, and discuss the pharmacogenomics of cancer therapy and its role in clinical trials.
Cancer
In order to truly understand the importance of pharmacogenomics in cancer therapy, one must have a good understanding of carcinogenesis and cancer genetics. The current view of cancer is that a malignancy arises from a transformation of the genetic material of a normal cell, followed by successive mutations, ultimately leading to the uncontrolled proliferation of progeny cells.4 Tumorigenesis is thought to require four to six rate-limiting mutation events to occur in the lineage of a single cell.4–6 Based on this working theory of cancer progression, newer genetic models of tumorigenesis have emerged and enhanced our current understanding of cancer genetics. One of these models is the first genetic model for colorectal tumorigenesis by Fearon and Vogelstein (Figure 13–1).7
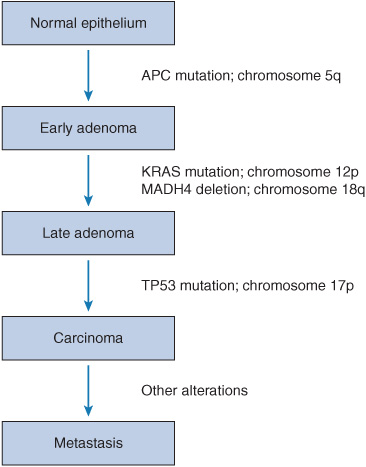
FIGURE 13–1 A genetic model for colorectal tumorigenesis. Tumorigenesis proceeds through a series of genetic alterations involving oncogenes (e.g., KRAS) and tumor suppressor genes (e.g., TP53 particularly those on chromosomes 5q, 17p, and 18q). (Adapted from Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. Jun 1 1990;61(5):759-767. Copyright 1990, with permission from Elsevier.)
The principles of Fearon and Vogelstein’s genetic model for colorectal tumorigenesis are as follows7: first, colorectal tumors arise as a result of the mutational activation of oncogenes coupled with the mutational inactivation of tumor suppressor genes (TSG); the latter changes predominate. Second, mutations in at least four to five genes are required for the formation of a malignant tumor. Third, although the genetic alterations often occur according to a preferred sequence, the total accumulation of changes, rather than their order, is responsible for determining the tumor’s biological properties. Fourth, in some cases, mutant TSG appear to exert a phenotypic effect even when present in the heterozygous state; thus, some TSG may not be “recessive” at the cellular level. V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS) is an oncogene involved in this process, and EGFR signal transduction results in KRAS activation. If KRAS is wild type, targeted therapies such as cetuximab (which is an EGFR inhibitor) are effective in the treatment of colon cancer. The general features of this model may be applicable to other common epithelial neoplasms.7
It is thought that alterations of cancer genes, resulting from gain-of-function mutations, can lead to overly active growth-promoting genes, which appear in cancerous cells as activated oncogenes.8 The precursors to these oncogenes, known as proto-oncogenes, are usually dominantly acting genes at the cellular level.9 The somatic mutations that cause activation of oncogenes are characterized by mutations that cause structural changes to the encoded protein, such as point mutations and chromosomal translocations.8 Proto-oncogenes can also be transformed to oncogenes by elevated expression through gene amplification or chromosomal translocations.10
In addition to the oncogenes, there are also TSG. These TSG operate to suppress cell proliferation through many biochemical mechanisms, and are often inactivated in various ways in cancer cells.11 An inherited mutant copy of a TSG increases susceptibility to specific types of cancer.10 The Knudson hypothesis of TSG inactivation postulates that mutant alleles of TSG are recessive at the cellular level.12 Therefore, both alleles of a TSG must be inactivated by loss-of-function mutations in the transformation of normal cells to cancerous cells. The loss of TSG function can occur by either genetic mutation or epigenetic silencing of genes via promoter methylation.13 Inactivation of TSG by mutation or methylation of one allele may be followed by other mechanisms that facilitate loss of the second copy,14 such as loss of heterozygosity (LOH) at the TSG locus.15
A census from the literature of reported cancer genes was recently compiled, which indicated that mutations in more than 1% of genes contribute to human cancer.9 Of the 291 reported cancer genes, 90% show somatic mutations that are acquired in cancer, 20% show germline mutations that predispose to cancer, and 10% show both (Figure 13–2). According to the census, the most common mutation class among the known cancer genes is a chromosomal translocation. Seventy percent of the cancer genes, altered by chromosomal translocations, are implicated in leukemias, lymphomas, and sarcomas but only represent 10% of human cancers.9 This bias is likely due to the fact that non-solid tumors such as leukemias are easier to analyze with cytogenetic techniques compared with solid tumors. Solid tumors have many translocations but few have been analyzed in great detail, except for sarcomas. The remaining 90% of cancers of epithelial origin have been shown to be altered by other types of mutations: base substitutions that lead to missense amino acid changes, nonsense changes, alterations in conserved splice site positions, and insertions or deletions in coding sequences or splice sites that may cause in-frame or frameshift alterations of the protein.

FIGURE 13–2 Schematic of the relative number of cancer genes with reported germline and somatic mutations in cancer. Adapted from The Biology of Cancer (© Garland Science 2007).10
GERMLINE VARIANTS AS PREDICTORS OF DRUG RESPONSE
Genetic variations are inherited DNA sequence mutations that typically cause minor physiologic changes, if any. Even with a complete loss of gene function due to a polymorphism, the biological impact is often modest. However, rare inherited DNA mutations known as germline mutations have shown to impact the risk of cancer, cancer prognosis, as well as treatment response and toxicity. Many of the first examples from pharmacogenomics in cytotoxic chemotherapy focus on understanding the inherited (germline) inter-individual differences involved in drug metabolism. Examples are described in Table 13–1.
TABLE 13–1 Examples of germline variants reported to modulate treatment outcomes.
TPMT and 6-Mercaptopurine
Thiopurine drugs, such as 6-mercaptopurine (6-MP), are purine antimetabolites used clinically to treat leukemias and are metabolized by the enzyme TPMT.16 Inter-individual variability in TPMT activity in red blood cells (RBCs) has been demonstrated to be a heritable autosomal co-dominant trait in exploratory studies.16 Over 11% of the population is found to have a coding nucleotide polymorphism, which is associated with low TPMT activity. More than 10% of the patients are heterozygous and only 0.3% of the population is homozygous for TPMT.16,17 The clinical importance of this variation was realized when patients who experienced severe hematological toxicity from thiopurine therapy were found to have low TPMP activity and elevated concentrations of mercaptopurine metabolites.18,19
TPMT is a cytosolic enzyme that preferentially catalyzes the S-methylation of aromatic and heterocyclic thio compounds such as 6-MP. 6-MP is an inactive prodrug and requires metabolism to 6-thioguanine (6-TG), which exerts cytotoxicity by incorporation into DNA and RNA20 (Figure 13–3). Alternatively, 6-MP undergoes inactivation by TPMT to thiouric acid and oxidation to 6-methylmercaptopurine by xanthine oxidase. However, hematopoietic cells (such as RBCs and leukocytes) do not have measurable xanthine oxidase activity, leaving TPMT as the major enzyme responsible for 6-MP inactivation.21 In the absence of or decreased TPMT enzyme activity, 6-MP is metabolized preferentially to 6-TG, which has potent bone marrow suppressive activity.
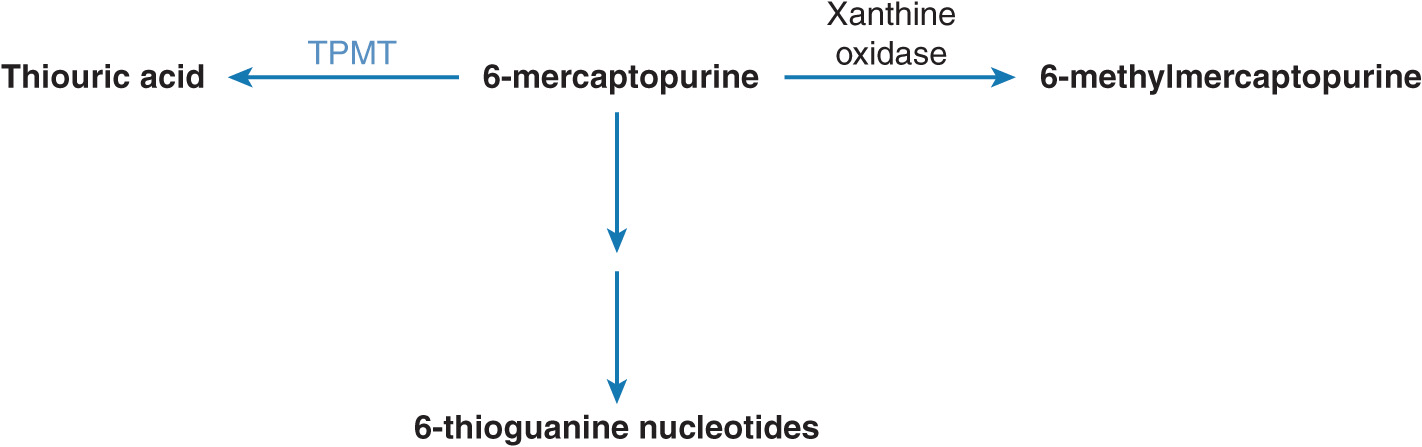
FIGURE 13–3 Metabolism of 6-mercaptopurine (6-MP). 6-MP is metabolically activated to form 6-thioguanine nucleotides. It undergoes metabolism catalyzed by xanthine oxidase or TPMT.
It is important to recognize such individuals both to avoid fatal bone marrow failure through inadvertent overdose and to ensure that an adequate drug effect can be achieved at a desirable dose.19,22 The potential benefit of testing TPMT activity to adjust the dose of 6-MP was demonstrated in a 6-year-old acute lymphoblastic leukemia (ALL) patient.23 After receiving a standard dose of 6-MP for post-remission therapy, the patient developed severe myelosuppression requiring a discontinuation of her treatment regimen. Interestingly, she was found to have 7× the population median value of 6-TG in her RBCs. Subsequent therapy with 6% of the standard 6-MP dosage allowed her to complete the potentially curative regimen without further toxicity.
Since assays measuring TPMT activity and RBC 6-TG concentrations are not clinically feasible, genetic testing has been developed to provide greater convenience and better reproducibility. This test evaluates three variant alleles known as TPMT*2, TPMT*3A, and TPMT*3C (Table 13–1), which accounts for 95% of the detected variant TPMT alleles. Importantly, there is also a high concordance between measured TPMT activity and the incidence of these polymorphisms.24,25 Genotyping for common TPMT alleles has shown to identify patients at risk of severe 6-MP toxicity such that TPMT genotyping can be integrated into the clinical management of patients undergoing 6-MP treatment.26 In July 2003, an advisory subcommittee of the FDA Center for Drug Evaluation and Research met to consider the role of TPMT genotyping in the administration of 6-MP treatment to pediatric leukemia patients. Even though comprehensive evaluation of the clinical benefit and cost-effectiveness of screening strategies with this test was not completed, the FDA decided that the evidence was sufficient to indicate benefit and to warrant informing prescribers, pharmacists, and patients of the availability of TPMT genotyping tests and their possible role in the selection and dosing of 6-MP.
There is now an FDA-approved genetic test (DNA-based) to determine which patients will be likely to experience severe myelosuppression prior to administration of 6-MP.27 However, in a recent cost-effectiveness analysis from a health care system perspective, screening for TPMT mutations via genotyping prior to the administration of 6-MP was not found to be cost-effective.28 In addition, some patients who developed severe myelo-suppression possessed normal TPMT activity. This suggests additional pathways are responsible for this toxicity.
In summary, measurement of TPMT genotypes and/or TPMT enzyme activity before instituting 6-MP may help prevent toxicity by identifying individuals with low or absent TPMT enzyme activity. Despite the usefulness of the TPMT genotyping test, it is recommended that clinicians monitor complete blood count (CBC) and liver function tests as well as clinical status routinely in patients undergoing 6-MP therapy. In addition, a normal TPMT enzyme activity in the screening test should not preclude this routine clinical assessment.
DPD and 5-Fluorouracil
5-FU is widely used as a part of combination chemotherapy regimen for the treatment of breast, colorectal, and head & neck cancers. Inside the tumor cells, 5-FU is bioconverted into cytotoxic nucleotides (Figure 13–4). The main mechanism of cytotoxicity is thought to be inhibition of thymidylate synthase by the metabolite 5-fluoro-2′-deoxyuridine-5′-monophosphate (5-FdUMP).29 Only a small part of the administered 5-FU dose exerts cytotoxicity since the majority of 5-FU is rapidly metabolized into inactive metabolites. The rate-limiting step is catabolism of 5-FU by DPD, which catalyzes the reduction of 5-FU into 5,6-dihydrofluorouracil (DHFU) (Figure 13–4). DPD plays a major role in the regulation of 5-FU metabolism and thus in the amount of 5-FU available for cytotoxicity.30–33
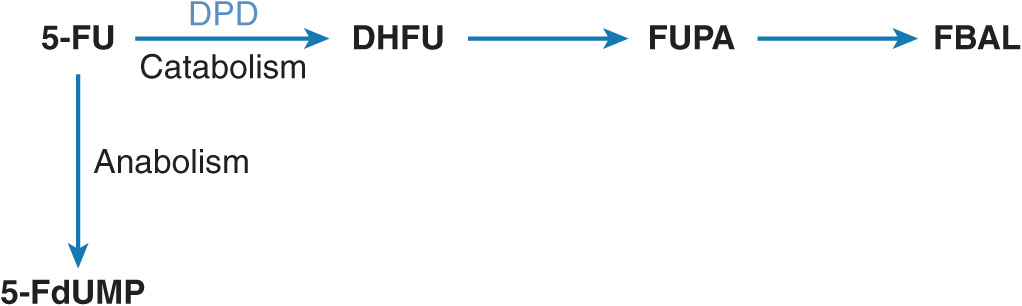
FIGURE 13–4 Metabolism of 5-FU. 5-Fluoro-2′-deoxyuridine-5′-monophosphate (5-FdUMP) is the active cytotoxic product resulting from a multistep 5-FU activation route. The initial and rate-limiting step is the catabolism of 5-FU by dihydropyrimidine dehydrogenase (DPD), catalyzing the reduction of 5-FU into 5,6-dihydrofluorouracil (DHFU). Subsequently, DHFU is degraded into fluoro-β-ureidopropionic acid (FUPA) and fluoro-β-alanine (FBAL).
In patients with DPD enzyme deficiency, 5-FU chemotherapy is associated with severe, life-threatening toxicity.34 Moreover, a markedly prolonged elimination half-life of 5-FU has been observed in a patient with complete deficiency of DPD enzyme activity.35 Several mutations in the dihydropyrimidine dehydrogenase gene (DPYD), which encodes for the DPD enzyme, have recently been identified.34,36 Population studies have suggested that 4–7% of the American population that exhibit a 5-FU dose-limiting toxicity might be associated with a genetic defect in the DPYD gene.37 Although complete DPD deficiency is rare, reduced levels of enzyme activity are more common, particularly in African Americans and women. These demographic differences have been illustrated in an analysis of enzyme levels from 258 normal volunteers. Partial DPD deficiency is present in 12.3% of black women, 4.0% of black men, 3.5 of white women, and 1.9% of white men.38
Many polymorphisms in the DPYD gene have been identified that may result in partial or total loss of DPD activity. A few alleles are associated with a marked decrease in DPD activity and enhanced fluoropyrimidine toxicity, the most important of which is DPYD*2A. In one series, at least one of three variants (DPYD*2A, DPYD*9B, or DPYD*13) has been found in 30% of 5-FU-treated patients (13 of 44) who developed grade 3 or 4 toxicities.39 This study demonstrated that patients with inherited high-risk DPYD variants have a 7-fold increased risk of severe toxicity with 5-FU. Other studies have found that DPYD*2A is present in less than 10% of patients with severe 5-FU toxicity. In one of these prospective studies of 683 patients receiving 5-FU monotherapy, genotyping has revealed the DPYD*2A allele in only 5% of those with treatment-related toxicities.40 Furthermore, less than one half of those with the DPYD*2A allele had grade 3 or 4 toxicity. Interestingly, the presence of DPYD*2A has been shown to be a stronger predictive factor for severe toxicity in men than in women.40
Inheritance of one of these alleles does not account for all cases of DPD deficiency. Impaired DPD activity has been detected in some patients with wild-type DPYD, presumably due to epigenetic mechanisms that regulate enzyme activity.41 To avoid the risk of severe and potentially fatal reactions, the manufacturers of both IV 5-FU and the oral form of fluoropyrimidine (e.g., capecitabine) recommend that these drugs be contraindicated in patients with known DPD deficiency. However, preemptive testing of all patients scheduled to receive 5-FU in order to identify those with a DPD deficiency is controversial and not widely practiced.
UGT1A1 and Irinotecan
Irinotecan is a topoisomerase I inhibitor used in combination with 5-FU as first-line therapy for the treatment of metastatic colorectal cancer.42 It is metabolized to its active metabolite, SN-38, which is 1,000× more potent than the parent drug (Figure 13–5).43 SN-38 is inactivated by a polymorphic hepatic enzyme UGT1A1.44 It has been observed that human liver samples harboring a seven dinucleotide TA repeat sequence (e.g., UGT1A1*28) in the UGT1A1 promoter have reduced SN-38 metabolism.44 Patients with the UGT1A1*28 allele have an increased risk for severe neutropenia, severe diarrhea, and death compared with patients without the UGT1A1*28 polymorphism.45
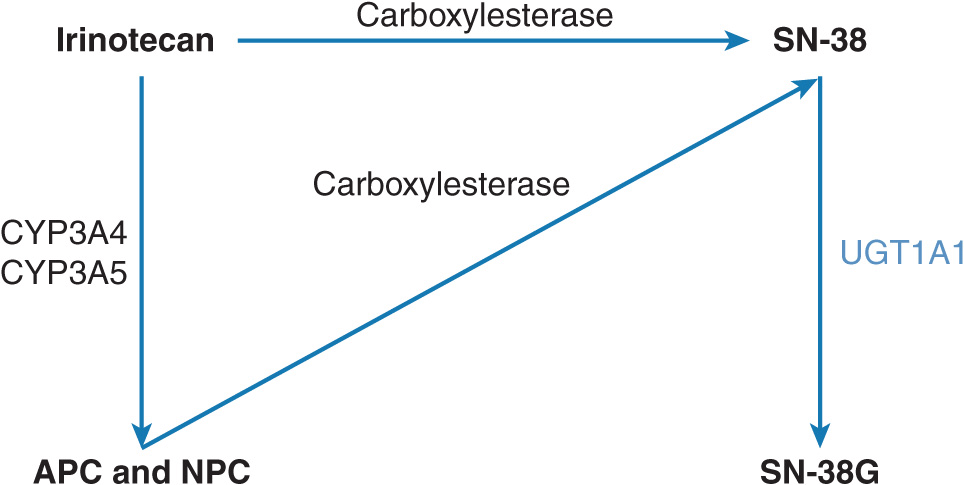
FIGURE 13–5 Metabolism of irinotecan. Irinotecan is primarily metabolized by human carboxylesterase 2 (hCE2) to the active metabolite, SN-38. SN-38 is glucuronidated by uridine diphosphate glucuronyl transferase (UGT1A1) to the inactive metabolite SN-38G, which is subsequently excreted. Other irinotecan metabolism pathways include oxidation by cytochrome P450 3A (CYP3A) into 7-ethyl-10[4-N-(5-aminopentanoic acid)-1-piperidino]-carbonyloxycamptothecin (APC) and 7-ethyl-10[4-(1-piperidino)-1-amino]-carbonyloxycamptothecin (NPC); SN-38 = 7-ethyl-10-hydroxy-camptothecin. In turn, NPC can also be converted into SN-38 by carboxylesterase.
Approximately 10% of the North American population is homozygous for the UGT1A1*28 allele (which is responsible for Gilbert syndrome); an additional 40% of the North Americans are heterozygotes (Camptosar package insert). In November 2004, the FDA Advisory Committee on Pharmaceutical Sciences considered the findings of several pharmacogenetic trials that had assessed the association between irinotecan-induced toxicities in patients who were homozygous with the UGT1A1*28/*28 genotype.45–48 These studies suggested that the UGT1A1*28/*28 genotype is associated with a decreased conversion of SN-38 to SN-38G, thus resulting in an increased severity of irinotecan-induced diarrhea and neutropenia.42,45,46 Based on these findings, the FDA advised an amendment of the product information to include information regarding the UGT1A1*28 polymorphism and hematological toxicity. In addition, the FDA recommended that patients with the UGT1A1*28/*28 genotype receive a lower starting dose of irinotecan. A pharmacogenomic test (i.e., Invader UGT1A1 Molecular Assay; Third Wave Technologies, Inc, Madison, WI) was FDA-approved to test for the UGT1A1*28 allele.
Recent data indicate that the association between UGT1A1*28 and irinotecan-induced neutropenia is dose-dependent. A meta-analysis showed that even though the inheritance of the UGT1A1*28/*28 genotype was associated with an increased risk of neutropenia at all doses, the relative risk for neutropenia at doses ≥250 mg/m2 was significantly higher (RR 7.0, 95% CI 3.10–16.78) than that for the lower doses (80–145 mg/m2 weekly, RR 2.43, 95% CI 1.34–4.39).49,50 This dose dependency on the risk of neutropenia could be explained by the relative extent of SN-38 glucuronidation. Similarly, the risk of severe diarrhea at medium and high doses of irinotecan was higher among patients with a UGT1A1*28/*28 genotype than among those with UGT1A1*1/*1 or UGT1A1*1/*28 genotypes.49 This increased risk of diarrhea in patients carrying UGT1A1*28 alleles was not apparent with lower doses (<125 mg/m2). In an evidence-based review, the quality of evidence on the analytical as well as clinical validity of current UGT1A1 testing methods was found to be adequate for severe toxicities such as neutropenia and diarrhea as stratified by UGT1A1 genotypes.51 The strongest association for a clinical end point was severe neutropenia since the patients homozygous for the UGT1A1*28 allele were 3.5 times more likely to develop severe neutropenia compared with individuals with the wild-type UGT1A1 genotype (risk ratio 3.51, 95% confidence interval 2.03–6.07). The cost-effectiveness of UGT1A1*28 genotyping has also been demonstrated primarily for Caucasian and African populations.52 These results show that UGT1A1 testing may be particularly useful in patients who are considered for medium- to high-dose irinotecan therapy to avoid severe neutropenia.
Despite strong evidence to support the use of UGT1A1 testing, routine use of this assay in all patients who are to receive irinotecan is not widely accepted. Even though such testing has been suggested to be cost-effective, the study lacked the assessment of the clinical relevance of identifying homozygotes. In addition, there are no current guidelines on how to dose the homozygous and heterozygous patients for the UGT1A1*28 allele(s). Additional clinical studies focusing on these key questions will facilitate implementation of UGT1A1 testing into clinical practice.
CYP2D6 and Tamoxifen
Tamoxifen is an anti-estrogenic drug used for the treatment of estrogen receptor–positive breast cancer.53 Although the estrogen receptor is a predictive marker of response to tamoxifen, not all women with estrogen receptor–positive breast cancer benefit from tamoxifen.54 Tamoxifen undergoes sequential biotransformation by several metabolic pathways (Figure 13–6).55,56 Endoxifen (4-hydroxy-N-desmethyl tamoxifen) is a more potent antiestrogen compared with tamoxifen as well as another active metabolite 4-hydroxytamoxifen (4-OH) in terms of relative contribution to the overall anti-cancer effect of tamoxifen. Tamoxifen to 4-OH is catalyzed by multiple enzymes. Cytochrome P-450 2D6 (CYP2D6) mediates oxidation of N-desmethyltamoxifen to endoxifen, which is also the most abundant tamoxifen metabolite.
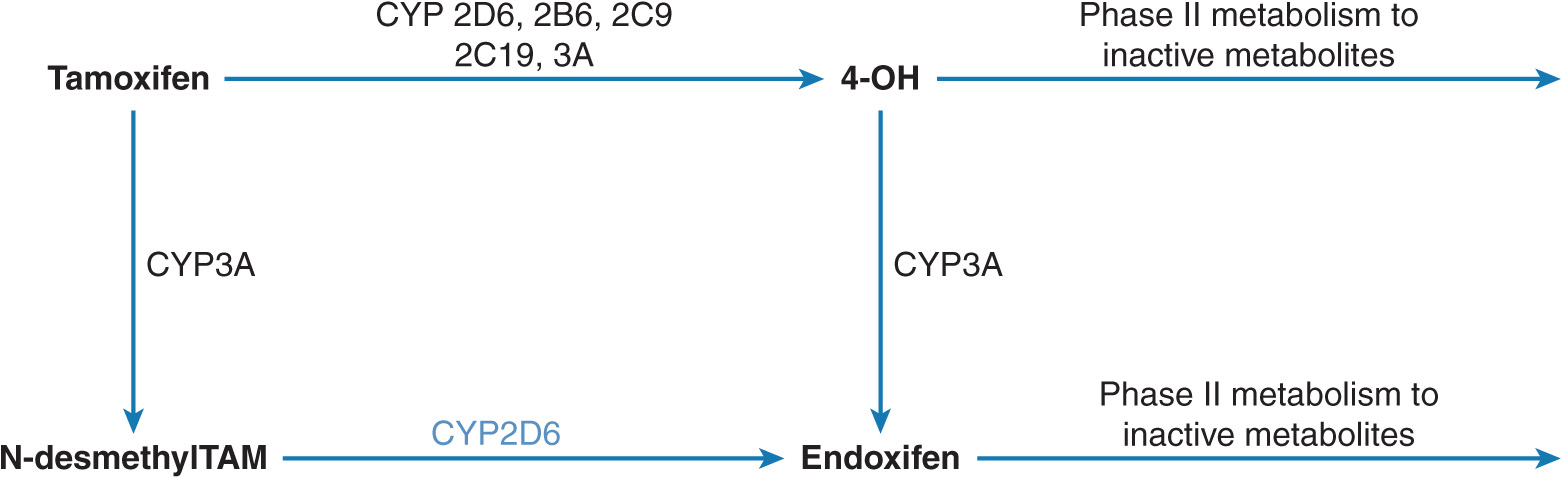
FIGURE 13–6 Biotransformation of tamoxifen to endoxifen. Tamoxifen is predominantly N-demethylated by the CYP3A enzyme to N-desmethyltamoxifen, which is the major primary metabolic pathway. This metabolite undergoes multiple oxidations including 4-hydroxylation by CYP2D6 to endoxifen. Tamoxifen 4-hydroxylation is metabolized by multiple CYPs to 4-hydroxytamoxifen (minor metabolic pathway). A small portion of endoxifen plasma concentrations appears to result from CYP3A-catalyzed N-demethylation of 4-hydroxytamoxifen. The hydroxylated metabolites undergo conjugation by phase II enzymes.
Initial clinical studies demonstrated that women on tamoxifen therapy who possessed genetic variants of CYP2D6 associated with low or no enzymatic activity or who received concomitant CYP2D6 inhibitors have significantly decreased endoxifen concentrations.57,58 Women possessing CYP2D6 variants responsible for decreased enzyme activity were found to have a higher risk of relapse and lower incidence of hot flashes.53,59 In addition, CYP2D6 metabolism was found to be an independent predictor of breast cancer outcome.53 Additional studies to confirm these results found that CYP2D6 poor and intermediate metabolizers had higher recurrence rates, all-cause mortality rates, and decreased event-free survival. Overall survival was not affected.58 Despite the positive findings in terms of the association between the CYP2D6 genotype and the effectiveness of tamoxifen in some studies, there are several studies that have not found a similar association.
This controversy was resolved by the recent retrospective analyses from two large clinical studies,60,61 which did not find an association between the CYP2D6 genotype and the effectiveness of tamoxifen in preventing breast cancer recurrence. In the Arimidex, Tamoxifen, Alone or in Combination (ATAC) trial, subjects who were genotyped (588 out of 3,116 women; 18%) and categorized into poor, intermediate, or extensive metabolizers of tamoxifen showed that the breast cancer recurrence rates were not significantly different in any of the groups in either the tamoxifen or the anastrozole (control) group.61 In addition, approximately 9% of women who were concomitantly taking potent CYP2D6 inhibitors (e.g., selective serotonin reuptake inhibitors [SSRIs]) did not have a significantly different clinical outcome compared with other women. Similarly, the analysis of the Breast International Group (BIG) 1-98 trial, involving 48% of the original study subjects (1,243 of 2,459 women) who were CYP2D6 genotyped and categorized into poor, intermediate, and extensive metabolizer groups, did not find any significant difference in event-free survival between groups.60 In addition, the incidence of hot flashes, which was previously associated with response, was not significantly different between groups. Although these studies were retrospective analyses and did not consider adherence to tamoxifen or the concomitant use of over-the-counter drugs, the conclusions provide evidence not to routinely recommend CYP2D6 testing in patients being considered for tamoxifen therapy. Even though the use of tamoxifen and strong CYP2D6 inhibitors such as SSRIs is controversial, the current recommendations to avoid SSRIs while on tamoxifen therapy are reasonable.
SOMATIC VARIANTS AS PREDICTORS OF DRUG RESPONSE
Somatic variations are acquired alterations in DNA that occur in cells other than the germ cells and are not inherited. Some of these mutations, detected in tumor DNA, predict response to cancer chemotherapy. The elucidation of the signal transduction networks that drive neoplastic transformation has led to rationally designed cancer therapeutics that target specific molecular events.62 These targeted therapeutics, unlike traditional cancer chemotherapeutics, do not have narrow therapeutic indices. Many of the currently known drugs in this class are protein kinase inhibitors. Genes that encode protein kinases are often dysregulated and constitutively activated in cancer. Kinase inhibitors therefore reduce the activity of the activated protein kinases, reducing the cellular oncogenic drive and inducing tumor regression. Three classic examples of somatic mutations of tumor DNA that predict response to kinase inhibitors are highlighted in Table 13–2.
TABLE 13–2 Examples of somatic mutations in target genes reported to modulate response to kinase inhibitors.

BCR-ABL, KIT, PDGFRA Kinase Inhibitors
The BCR-ABL protein tyrosine kinase is the fusion of the BCR and non-receptor protein tyrosine kinase ABL that results from the reciprocal chromosomal translocation t(9;22) producing a shortened chromosome 22, called the Philadelphia (Ph) chromosome. This resultant chromosome has constitutive tyrosine kinase activity.63 The BCR-ABL protein is associated predominantly with CML but also with ALL.63 Imatinib was the first small molecule kinase inhibitor approved as treatment for CML that targets BCR-ABL. It binds to an inactive conformation of the BCR-ABL protein kinase.64,65 Imatinib also has specificity for the platelet-derived growth factor receptor alpha (PDGFRA) and stem cell growth factor receptor (KIT) protein kinases, and is used in treatment of malignancies associated with dysregulated forms of those proteins.64
Treatment of CML patients with imatinib leads to complete cytogenetic and hematological remission; however, imatinib fails to deplete leukemic stem cells that harbor the BCR-ABL fusion protein.66 Therefore, some patients develop resistance to imatinib, particularly in the advanced phases of CML and Ph-positive ALL.67 Mechanisms of imatinib resistance involve BCR-ABL amplification and overexpression of mRNA and protein.68,69 However, the most common mechanism of resistance is the acquisition of point mutations in the kinase domain of the ABL gene.70
Point mutations in the ABL gene are listed in Table 13–3. The first identified point mutation associated with imatinib resistance was T315I.70 Crystal structures of an analogue of imatinib bound to the ABL kinase domain revealed that the T315 residue was crucial for the interaction between imatinib and ABL.71 It was also found that a different mutation at the T315 residue (T315V) conferred constitutive kinase activity to ABL and was less sensitive to imatinib compared with wild-type ABL.72 The T315I point mutation impairs imatinib binding, thereby reducing tyrosine kinase inhibition of ABL. More than 50 different point mutations in ABL associated with imatinib resistance have been reported.67 Most are rare, and six amino acid residues (G250, Y253, E255, T315, M351, and F359) thus far account for 60–70% of imatinib-resistant mutations.67 Two of the more frequently detected ABL mutants, Y253F and E255K, have been shown to have high in vitro transforming potential. The in vitro finding is consistent with clinical findings that show P-loop mutations such as Y253F and E255K are associated with a greater likelihood of progression to blast crisis and decreased overall survival in imatinib-treated patients.73 Similarly the T315I mutation, generally found in patients with advanced CML, has a worse overall survival compared with other ABL mutations in patients on imatinib therapy.74 This is likely due to the fact that no FDA-approved tyrosine kinase inhibitors are effective against the T315I mutation. Current standard of care for patients with the T315I mutation is to enroll them in a clinical trial or refer them for stem cell transplant.
TABLE 13–3 Activity of BCR-ABL inhibitors against a selection of BCR-ABL1 mutants found in patients with CML.
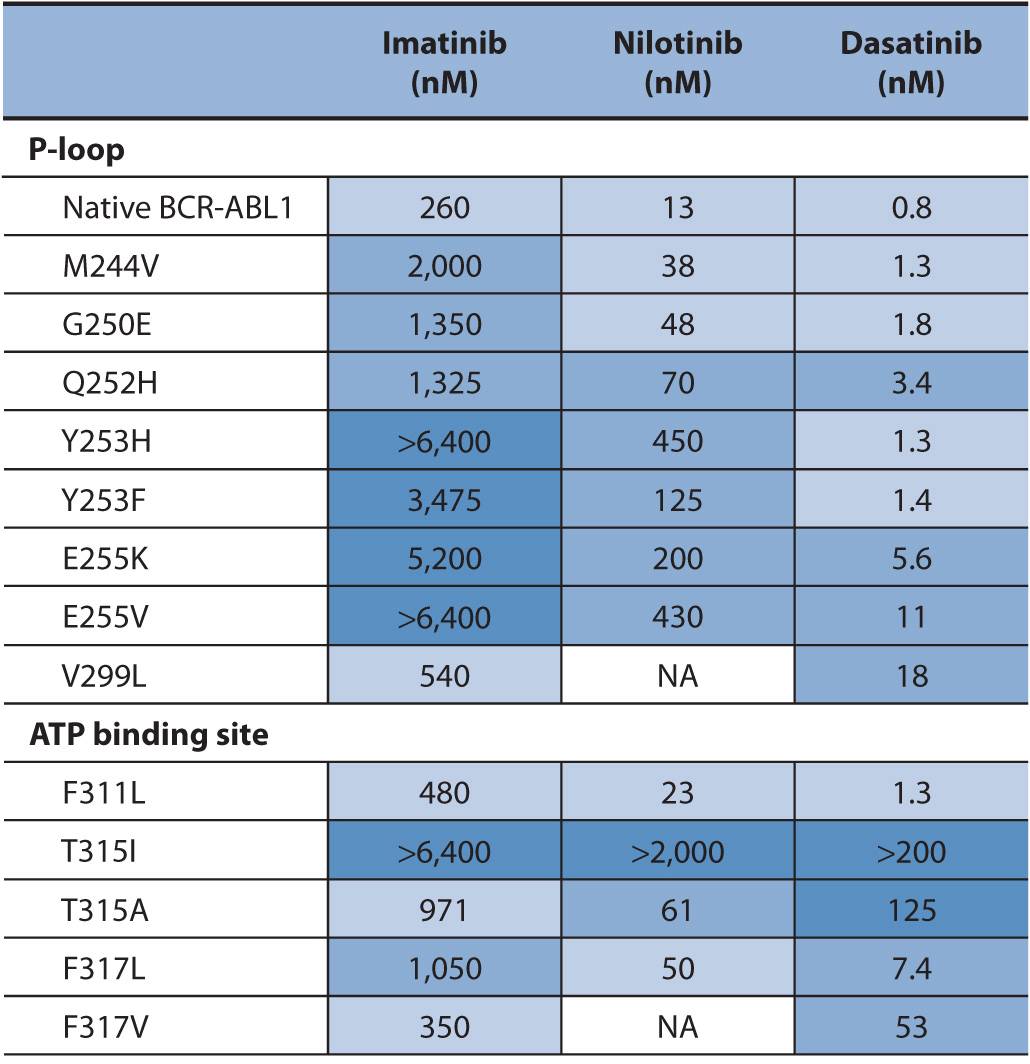
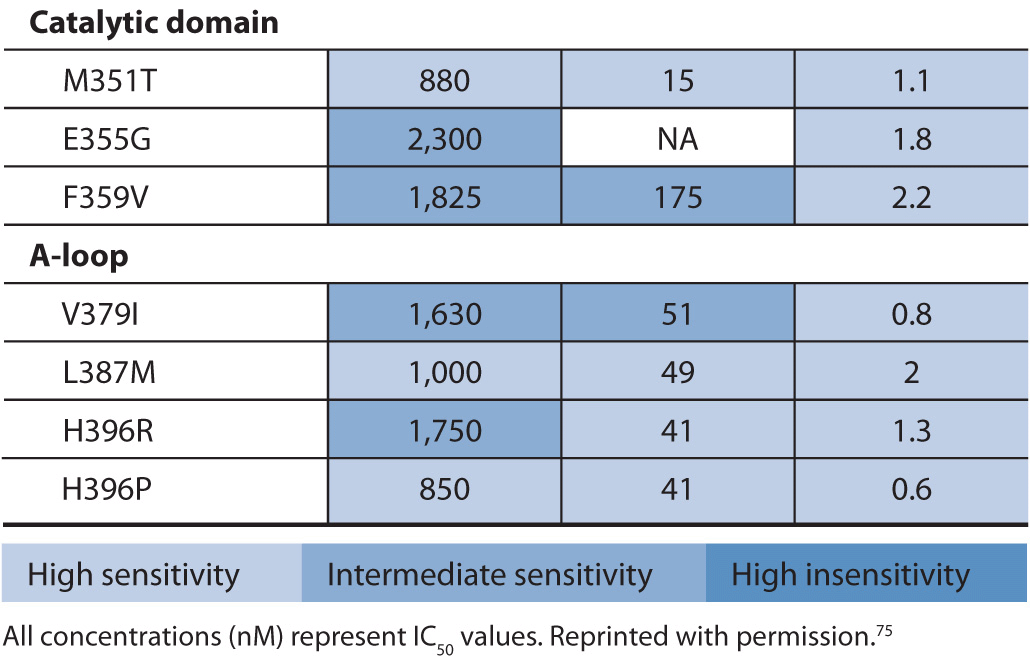
Drug-resistant BCR-ABL point mutations are found in imatinib-naïve CML or are acquired during imatinib treatment. Acquired imatinib resistance involves the re-emergence of BCR-ABL tyrosine kinase activity. This suggests that the mutant BCR-ABL protein is still a viable target for inhibition by small molecule inhibitors.76,77 To this end, alternative therapies have been designed to overcome resistance to imatinib. One of these drugs, nilotinib, is approximately 30-fold more potent than imatinib as an ABL inhibitor.67 A significant clinical response to nilotinib is demonstrated in patients with imatinib-resistant CML in all phases of disease and in patients with Ph-positive ALL.78 Nilotinib also inhibits KIT and PDGFRB protein kinases.79
Another drug designed to overcome imatinib resistance is dasatinib. Dasatinib is a potent inhibitor of BCR-ABL, Srcfamily kinases, KIT, and PDGFR.80,81 In contrast to imatinib and nilotinib, dasatinib binds to the active conformation of the ABL kinase.82 Based on data from phase I and II trials in patients with imatinib-resistant CML and Ph-positive ALL patients, dasatinib is approved in the United States for the treatment of adults in all phases of CML with imatinib resistance or intolerance and in patients with Ph-positive ALL with imatinib resistance or intolerance.83,84 The sensitivity of the point mutations in BCR-ABL to imatinib, nilotinib, and dasatinib is provided in Table 13–3. Clinically, the assessment of these point mutations is only done if a patient is not responding to therapy.
Most gastrointestinal stromal tumors (GISTs) harbor oncogenic KIT or PDGFRA receptor tyrosine kinase mutations.85 The KIT or PDGFRA gain-of-function mutations are early events in GIST oncogenesis.86 Imatinib, a potent inhibitor of KIT signaling, has recently become first-line treatment of metastatic GIST following in vitro studies suggesting a therapeutic potential for imatinib in a human GIST cell line.87 Prior to imatinib, surgical resection of primary localized GIST was the only chance for cure.88 GISTs are refractory to standard chemotherapy and radiation, with a predicted 5-year survival of 30%.89 Prospective trials of imatinib in metastatic GIST have shown that approximately 80% of patients respond to imatinib or have stable disease.90,91 In addition, 70% of metastatic GIST patients will have at least a 2-year disease-free survival and 50% will be free of disease progression.92
KIT mutations occur in up to 90% of GISTs and clinical response to imatinib is dependent on the presence of specific KIT mutations.93 Exon 11 KIT mutations are found in 75% of GISTs and result in the abrogation of the juxtamembrane auto-inhibition of KIT kinase.94 Patients with exon 11 KIT mutations have a higher response rate to imatinib treatment and longer time to treatment failure compared with patients with other KIT or PDGFRA mutations.95 In addition, patients with the rare exon 13 KIT mutation or exon 17 PDGFRA mutation may respond to imatinib.95 Patients without any detectable KIT or PDGFRA mutations respond less frequently to imatinib treatment compared with patients with exon 11 KIT mutations.93 However, only up to 39% of those patients without KIT or PDGFRA mutations do respond to imatinib.96 These data suggest that imatinib treatment ought to be considered for all GIST patients, regardless of KIT or PDGFRA mutation status.95 The only exception may be patients with a primary imatinib-resistant mutation of PDGFRA (D842V).
The majority of metastatic GIST patients will develop resistance to imatinib. The most common resistance mechanisms involve the acquisition of secondary exon 13, 14, or 17 KIT mutations that prohibit imatinib binding.95 Some of these secondary mutations, such as the frequently occurring V654A substitution, are intrinsically imatinib-resistant.97 However, other mutations such as those involving the N822 residue are intrinsically imatinib-sensitive but are associated with clinical imatinib resistance when coincident with an exon 11 KIT mutation.97
Sunitinib, an inhibitor of KIT, PDGFRA, FLT3, and VEGFR2, has recently been approved for the treatment of imatinib-resistant GIST and in patients unable to tolerate imatinib therapy.98
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


