Pharmacogenomics in Neurology
LEARNING OBJECTIVES
INTRODUCTION
Knowledge of pharmacogenomics is becoming increasingly important in neurology, as in many fields of medicine, for practicing pharmacotherapeutics. The effectiveness and toxicities of medications used in secondary stroke prophylaxis, dementia, seizure disorders, multiple sclerosis (MS), and Parkinson disease (PD) are all influenced by genetic polymorphisms. These include but go beyond variability in metabolism by Phase I/II enzymes. Genetic variability plays a role in how prodrugs work by controlling metabolism to their active form, as in the case of clopidogrel; how drugs bind to receptors, as in the case of MS; or how drugs are eliminated or inactivated as in the case of anticonvulsants and agents for Alzheimer’s disease (AD).1 In recent AD clinical trials, for example, it has been found that a certain apolipoprotein E (APOE) allele can affect whether patients respond positively to a drug treatment. Susceptibility to life-threatening hypersensitivity reactions is also related to specific polymorphisms. Future research may provide genetic explanations for multiple drug resistance in epileptic patients.
ANTIPLATELET AGENTS FOR STROKE PROPHYLAXIS
Antiplatelet therapy is the standard of care for prevention and treatment of atherothrombotic events, including myocardial infarction and stroke prophylaxis.2 Variability in antiplatelet response to all classes of these drugs has been well documented and lack of response to antiplatelet therapy has been established as a risk factor for developing secondary atherothrombotic events. While pharmacokinetics and disease severity play a role in the variability in response to antiplatelet agents, genetic polymorphisms contribute to variability in response to aspirin, the thienopyridines, and the GPIIb/IIIa inhibitors. Patients with certain genotypes may benefit from individualized therapy to reduce their risk of secondary thrombotic events.
Thienopyridines
Clopidogrel is commonly used and indicated for acute myocardial infarction, myocardial infarction prophylaxis, stroke prophylaxis, and percutaneous coronary intervention.3 It is administered as an inactive drug that requires activation, largely by CYP2C19, CYP3A4 and CYPA12, to an active metabolite for a therapeutic effect to be produced (Figure 16–1). Antiplatelet effect is produced by the active metabolite of clopidogrel irreversibly inhibiting adenosine diphosphate (ADP)–induced platelet aggregation by preventing ADP binding to its P2Y12 platelet receptor. It is estimated that up to 40% of patients do not achieve an optimal antiplatelet effect with clopidogrel, which puts these patients at higher risk of developing an atherothrombotic event.4
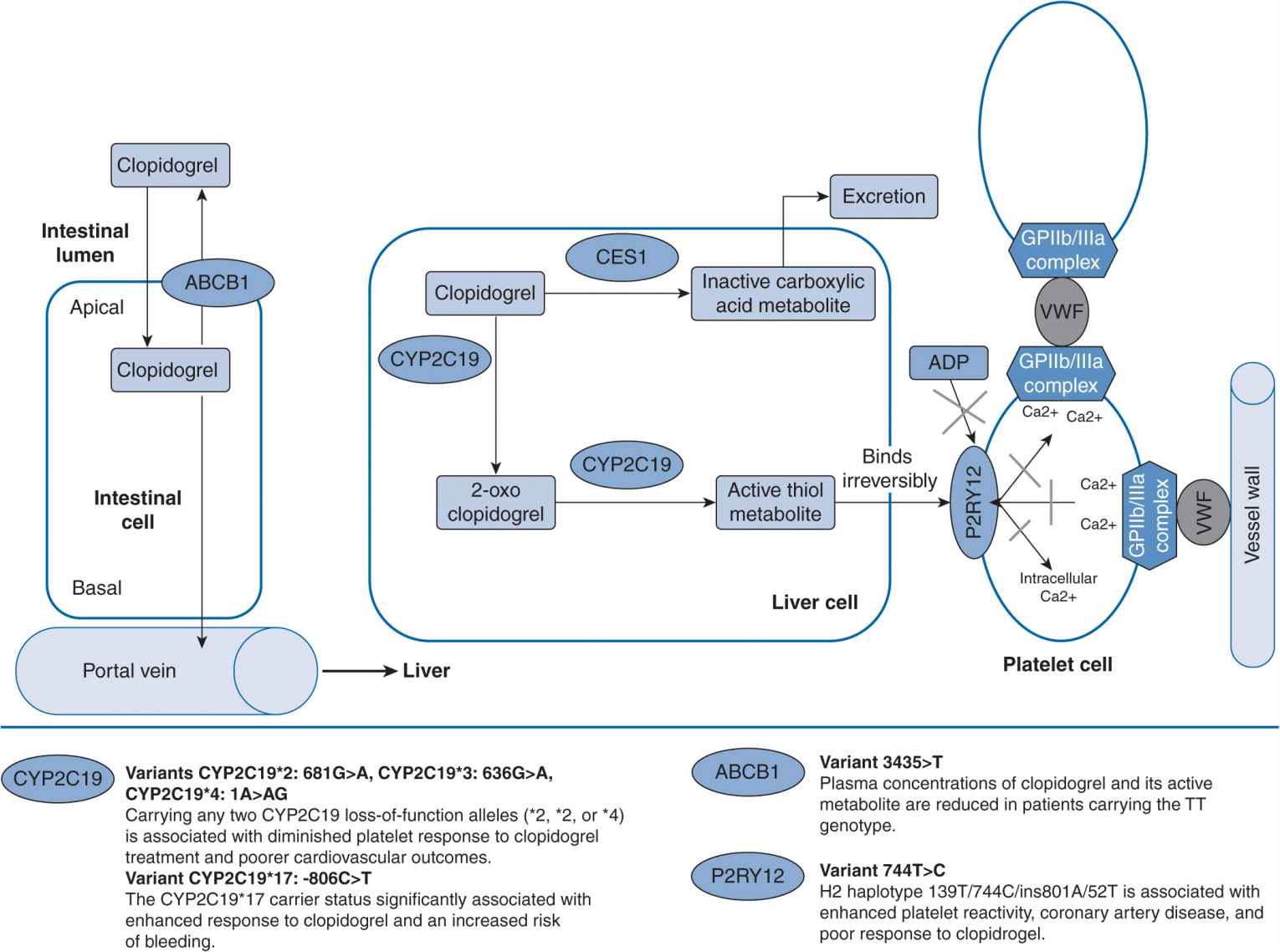
FIGURE 16–1 Genetic polymorphisms associated with clopidogrel.127 (With permission from Vance JM., Tekin D. Genomic medicine and neurology. Continuum: lifelong learning in neurology. Neurogenetics. 2011;17(2):249–267. Copyright © 2011, American Academy of Neurology).
Mega et al. tested the association between genetic variants in CYP genes and antiplatelet response in 162 healthy patients taking clopidogrel.5 They additionally examined the association between these genetic variants and cardiovascular outcomes in a cohort of 1,477 subjects. Patients with a reduced function allele for CYP2C19 had a relative reduction of 32.4% of the active metabolite exposure compared with patients without a reduced function allele (P <.001). The ex vivo antiplatelet effect measured in these patients was 25% less than that in patients without the reduced function allele. Patients with the ultrarapid metabolizer (UM) genotypes had the highest concentration of active metabolite and the greatest platelet inhibition, and subjects with poor metabolizer (PM) genotypes had the lowest concentration of active metabolite and the lowest platelet inhibition. A similar trend was observed for patients with a reduced function allele for CYP2B6. There was no association of reduced function alleles for CYP2C9, 3A5, and 1A2 and production of the active metabolite of clopidogrel or in antiplatelet response. Patients with at least one reduced function CYP2C19 allele were at a significantly higher risk for death due to cardiovascular causes, myocardial infarctions, or stroke compared with patients with a normal functioning CYP2C19. The most common reduced function allele was CYP2C19*2. There was no an association between the primary efficacy outcome and any of the other CYP genotypes. Patients with CYP2C19*2 had significantly lower concentrations of active metabolite of clopidogrel, decreased platelet inhibition, and a higher rate of major cardiovascular events. These findings have been confirmed in numerous other studies.6–8
Other reports have investigated the response variability to clopidogrel in patients with genetic polymorphisms CYP2C19*2, *3, and *17, CYP3A4*1B, and CYP3A5*3.9 Previous results show that CYP2C19*2 allele carriers had significantly increased platelet aggregation. There were no other associations between the other genotypes and platelet aggregation. This has been replicated in other studies8,10 by Mega et al.5 and Simon et al.8
Some individuals may have an increased response to clopidogrel, which would increase their risk of bleeding complications. Data from a retrospective study of 598 patients after the administration of 600 mg of clopidogrel demonstrated that the CYP2C19*17 genotype was associated with extensive metabolism of clopidogrel and better platelet response to clopidogrel, while the *4, *5, and *6 alleles did not produce significant effects. The *17 allele was associated with an increased risk of bleeding.10 Prospective studies by Geisler et al.9 and Mega et al.5 failed to find an association between patients with the *17 allele and an altered response to clopidogrel, but Frere et al.10 and Sibbing et al.16 found that patients with this allele may have a better antiplatelet response to clopidogrel. Further, findings from Sibbing et al. showed that these patients were at an increased risk of bleeding. Considering the implications of putting patients at a higher risk of bleeding, genetic testing may be appropriate to determine which patients have this gain-of-function allele, specifically for patients already at high risk for bleeding. More clinical studies are needed to clarify the effects of having a CYP2C19*17 allele when taking clopidogrel.
Although Figure 16–1 does not include the CYP1A2 enzyme, it has been shown to influence the production of the active metabolite of clopidogrel. Medications that induce the CYP1A2 enzyme have been shown to enhance the antiplatelet effects of clopidogrel. These would include insulins, modafinil, some beta-lactam antibiotics, and tobacco. There have been several reanalyses of large studies including CAPRIE, CURE, CREDO, and CLARITY TIMI. Endpoints for CAPRIE included stroke but the endpoints for the others were death or coronary events. The reanalysis stratified patients as active smokers vs nonsmokers or former smokers. Each of these analyses demonstrated that clopidogrel was much more efficacious in smokers compared with nonsmokers, and attributes this finding to the induction of the CYP1A2 enzyme in smokers.11
It is clear that genomic polymorphisms are associated with variability in response to clopidogrel. This is more relevant when clopidogrel is used in the setting of acute stroke than in the setting of acute coronary syndrome. In acute coronary syndrome a loading dose of 300–600 mg is often used that would at least temporally avoid the problem of inadequate production of the active thiol metabolite. However, because of the risk of hemorrhagic transformation of a cerebral infarction, loading doses are not commonly used following an acute stroke. The polymorphisms associated with reduced clopidogrel metabolism can and do significantly limit its effectiveness particularly in the days following an acute stroke when the risk of reinfarction is the highest.
Aspirin
Aspirin has remained the gold standard for secondary stroke prophylaxis.12 Acetylsalicylic acid acetylates a serine moiety on the COX-1 enzyme rendering it incapable of metabolizing arachidonic acid to thromboxane, a potent agonist for platelet aggregation. Because platelets are without a nucleus and incapable of protein synthesis, the COX-1 enzyme cannot be regenerated, and the platelets’ ability to aggregate is permanently disrupted. The effects of a single dose of aspirin reverse only as the population of platelets is turned over and replaced by new platelets. Acetylsalicylic acid is the only salicylate that affects platelet aggregation, and it is deacetylated rapidly, largely in the portal circulation. Many studies have reported “aspirin resistance.” Our own series reports an inadequate antiplatelet response to aspirin in 20% of patients being treated for secondary stroke prophylaxis.
Patients resistant to 81 mg do respond to higher aspirin doses. However, there are a substantial number of patients who do not achieve an antiplatelet effect at any aspirin dose. There are some data to suggest that genomic polymorphisms may contribute to this lack of responsiveness to aspirin. Halushka et al. studied 38 healthy volunteers to assess if genetic variation of COX-1 influenced response to aspirin.13 They performed genomic sequencing of the patients’ cDNA of COX-1 and measured platelet aggregation as well as production of thromboxane B2 and prostaglandin F2∞. After genomic sequencing, nine different polymorphisms of COX-1 were identified. Four of these polymorphisms were common, with heterozygosity greater than 10%, and included A-842G, C50T, C644A, and C22T. Two of these polymorphisms, A-842G and C50T, were found to be in complete linkage disequilibrium. These polymorphisms were further evaluated for association with activity of COX-1. There were no significant differences in thromboxane B2 production found between homozygotes for the common alleles and heterozygotes for any of these polymorphisms. Response to aspirin as measured by formation of prostaglandin F2∞ was decreased in heterozygous patients for A-842G and C50T versus the common allele homozygous patients indicating that patients with this genetic variant in COX-1 may have an increased sensitivity to aspirin. There were no significant differences found in the response to aspirin for patients with the C644A or C22T polymorphisms. Platelet aggregation following aspirin showed a reduced response in patients homozygous for the common allele of A-842G and C50T, compared with that in heterozygous patients. These data represent a starting point for future in vivo studies to build upon to determine the significance of the A-842G and C50T genetic variant and aspirin response.
These results, however, conflict with subsequent work of others who report that the COX-1 haplotypes were associated with different antiplatelet response. There was an association between haplotype GCGCC, including A-842G, and increased platelet aggregation and thromboxane production (i.e., decreased sensitivity to aspirin).14
It has also been postulated that a polymorphism in glyco-protein IIIa may influence aspirin response variability. Szczeklik et al. investigated the effect of the polymorphism T1565C (Leu33Pro, P1A1/A2) of glycoprotein IIIa on response to aspirin in healthy men.15 Bleeding time was shorter before aspirin ingestion in patients with the P1A2 allele and aspirin prolonged bleeding time in both groups. In 7 out of 26 patients with the P1A2 allele aspirin shortened bleeding time by 30 seconds, compared with 1 out of 54 patients with the common allele. While bleeding time is not a precise measure of the effects of aspirin, there does seem to be an association between P1A2 allele and the reduced response to aspirin. The P1A1/A2 and P1A2/A2 polymorphisms have been studied by others.16 Patients with P1A2 were significantly more sensitive to the platelet inhibition by acetylsalicylic acid than P1A1 patients, contradicting Szczeklik’s17 results that indicated patients with the P1A2 allele were more resistant to platelet inhibition by acetylsalicylic acid.
These conflicting results may have been due to technical differences in the way that response to aspirin was measured or because platelets were incubated with aspirin rather than administering aspirin to the subject. Mechanisms to explain “aspirin resistance” are varied including pharmacokinetic, disease, and environmental influence and now the beginnings of data to suggest genetic factors.
GPIIb/IIIa Inhibitors
GPIIb/IIIa inhibitors are a class of antiplatelet drugs that are largely used for percutaneous coronary intervention and myocardial infarction prophylaxis and include abciximab, tirofiban, and eptifibatide.17 They act by inhibiting glycoprotein IIb/IIIa and preventing the attachment of adhesive ligands to the glyco-protein IIb/IIIa receptor on activated platelets. These drugs also have a documented response variability, which may put patients at risk for cardiovascular events.
It has been suggested that patients with a P1A2 polymorphism may have an altered response to GPIIb/IIIa inhibitors, such as abciximab.18 Differences were found in inhibition of platelet aggregation among different genotypes, with heterozygotes having a lower IC50 than homozygotes for P1A2. The IC50 for heterozygote patients was 1.9 ± 0.21 μg/mL, compared with 2.27 ± 0.19 μg/mL for patients homozygous for A1 and 2.13 ± 0.14 μg/mL for patients homozygous for A2. It appears that patients heterozygous for P1A2 were more sensitive to the inhibition of platelet aggregation by abciximab.
Conclusions
Based on the studies reviewed, it was shown that clopidogrel, aspirin, and GPIIb/IIIa inhibitors all have genetic variability that could influence antiplatelet response. There are the most data for clopidogrel, and these studies largely support the claim that individuals with particular polymorphisms will have an altered response to the antiplatelet effect of clopidogrel. The genotype with the most supporting data is for individuals expressing a CYP2C19*2 allele, which is a loss-of-function allele. Numerous trials have shown that individuals carrying this allele have a reduced amount of the active metabolite of clopidogrel, lower platelet inhibition, and an increased risk for cardiovascular events.5–8 Considering these significant results, genetic testing may play an important role in determining patient selection for clopidogrel. In fact, in May 2009 the FDA added a warning to the Plavix package insert stating that PM of CYP2C19 are at a higher risk of developing cardiovascular events and that genetic testing is available to determine patients who are PM.3 Additionally, they recommended that alternative treatment be used in these patients. The evidence for an altered response to clopidogrel in patients with a CYP2C19*17 allele, a gain-of-function allele, is not as clear and more clinical studies are warranted, particularly considering the implications of increased bleeding.
There are also conflicting data on the role of genetic polymorphisms in controlling aspirin response variability. The most common polymorphisms studied have been polymorphisms of COX-1, specifically A-842G/C50T which are in linkage disequilibrium. Halushka et al. found that patients with an A-842G/C50T polymorphism may have an increased sensitivity to aspirin, as patients with this polymorphism had an increased inhibition of formation of prostaglandin H2 by acetylsalicylic acid compared with patients with the common allele.13 Maree et al., conversely, showed that patients with an A-842G/C50T allele actually had a decreased sensitivity to aspirin.14 Together, these studies show that more pharmacogenomic research is required to clarify the effects of this polymorphism on aspirin response.
A polymorphism in glycoprotein IIIa, P1A1A2, has also been linked to variations in aspirin response as well as GPIIb/IIIa inhibitors response. Two trials in healthy volunteers studied the effect of this polymorphism on bleeding time and platelet aggregation after treatment with aspirin.15,18 They produced conflicting results, with Szczeklik et al.15 showing that patients with this polymorphism had a decreased sensitivity to aspirin due to shortened bleeding time and Andrioli et al.18 finding that patients with this polymorphism had an increased sensitivity to platelet inhibition by aspirin. Additionally, conflicting results were also produced when patients with this polymorphism were treated with GPIIb/IIIa inhibitors. An in vitro study by Michelson et al.19 found these patients to have an increased sensitivity to abciximab, whereas Weber et al.20 could not show any significant difference. Therefore, it cannot be concluded that patients with the P1A1A2 polymorphism have an altered response to either aspirin or GPIIb/IIIa inhibitors, although this may be the case. Future studies should focus on this polymorphism to determine if any association can be made with response variability. At this time, scientific evidence does not support a recommendation for genetic screening to identify individuals with this polymorphism before receiving aspirin prophylaxis.
ANTICOAGUALANTS IN STROKE PROPHYLAXSIS
Atrial fibrillation is a common disorder that increases the risk of ischemic stroke. The incidence of atrial fibrillation in 2007 was more than 5% of individuals over the age of 65 in the western world.21 Warfarin, a vitamin K antagonist, has been the mainstay therapy to prevent such related thromboembolism. It has long been thought of as a difficult drug to manage as a result of its narrow therapeutic index in that toxic doses are not significantly higher than effective doses. However, warfarin is also difficult to dose due to genetic variants in the CYP2C9 and VKORC1 genes (Figure 16–2). Patient responses to warfarin’s anticoagualant effects are highly variable. Warfarin is a racemic mixture of R– and S-warfarin, with S-warfarin being the more potent of the two isomers. S-Warfarin is predominantly metabolized by the CYP2C9 enzyme. The major variants of CYP2C9 are the *2 and *3 alleles, and they are associated with reduced enzyme function and thus slower clearance resulting in higher drug concentrations. Patients with the CYP2C9*2 and CYP2C9*3 variants, when compared with patients with wild genotype CYP2C9*1/*1, took a longer time to reach therapeutic international normalized ratio (INR), had an increased rate of above range INR and increased risk of bleeding, and required a lower maintenance dose of warfarin.22 VKORC1 is the target enzyme of warfarin, and VKORC1 haplotypes A and B account for 25% of warfarin dose variance.23 Patients with haplotype A require a lower warfarin dose, whereas those with haplotype B require a higher warfarin dose. The FDA has provided specific dosing recommendations for those with CYP2C9 and VKORC1 variants as of January 2010. The recommended ranges for maintenance doses for genetic variants are included in the Coumadin package insert, and should be considered on dose initiation.24
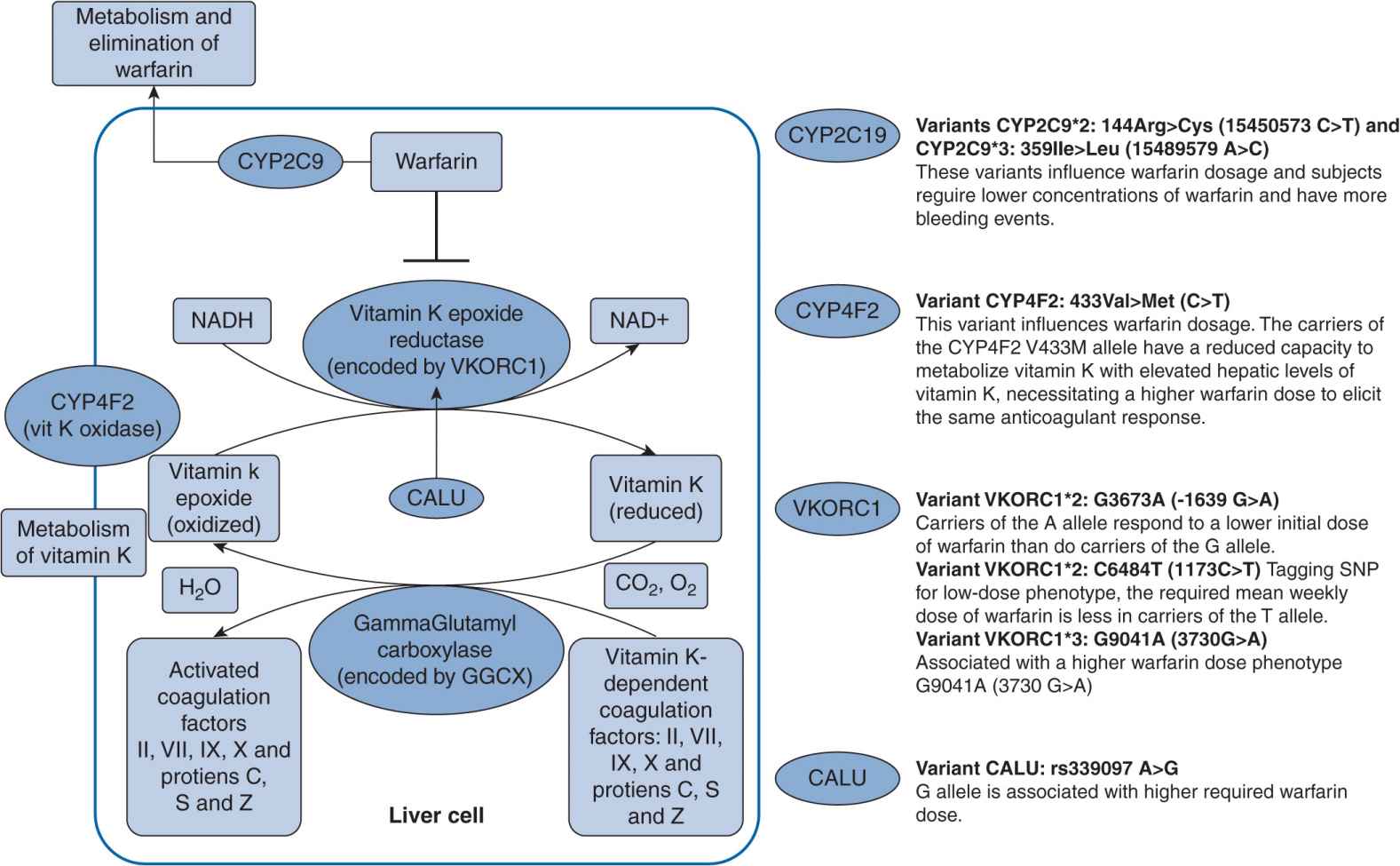
FIGURE 16–2 Genetic polymorphisms associated with warfarin.127 (With permission from Vance JM., Tekin D. Genomic medicine and neurology. Continuum: lifelong learning in neurology. Neurogenetics. 2011;17(2):249–267. Copyright © 2011, American Academy of Neurology).
Schwarz et al. conducted a prospective study to evaluate the CYP2C9 and VKORC1 contributions to initial (the first 4 weeks of therapy) variability in INR response. CYP2C9 and VKORC1 genotyping was performed, and major variants, such as CYP2C9*2 and CYP2C9*3 genotypes and VKORC1 A and non-A haplotypes, were identified.25 The allelic frequencies of CYP2C9*2, CYP2C9*3, and VKORC1 haplotype A in the patients were 12.0%, 4.8%, and 32.6%, respectively. Clearly patients who were VKORC1 A/A homozygotes had a 2.4 times faster rate of achieving therapeutic INR than VKORC1 non-A/non-A homozygotes (P <.001). The A/A homozygotes also had a 2.5 times higher rate of achieving an INR greater than 4.0 than non-A/non-A homozygotes ![]() . Patients who were A/non-A heterozygotes were at higher risk as well. A/A homozygotes were above therapeutic INR for a longer period of time. Overall, they required a lower average dose of warfarin. CYP2C9 variants did not significantly affect the time to first therapeutic INR and time above an
. Patients who were A/non-A heterozygotes were at higher risk as well. A/A homozygotes were above therapeutic INR for a longer period of time. Overall, they required a lower average dose of warfarin. CYP2C9 variants did not significantly affect the time to first therapeutic INR and time above an ![]() . However, patients with CYP2C9 variants had a shorter time to a first
. However, patients with CYP2C9 variants had a shorter time to a first ![]()
![]() . In the initial stages of warfarin therapy, INR variations are contributed to VKORC1 haplotype A.
. In the initial stages of warfarin therapy, INR variations are contributed to VKORC1 haplotype A.
The International Warfarin Pharmacogenetics Consortium developed a pharmacogenetic dose algorithm for warfarin and to compare it with clinical dose and fixed-dose algorithms.26 Clinical and genetic data for 5,700 patients were obtained from 21 different research groups. Of particular interest were the patients’ demographics, concurrent medications, primary indication for warfarin, maintenance dose of warfarin, target INR, and CYP2C9 and VKORC1 genotypes. Combining clinical and genetic data, a pharmacogenetic dose algorithm was developed (available at www.warfarindosing.org). The results of the study show that the pharmacogenetic dose algorithm was more accurate in estimating doses than either clinical or fixed-dose algorithms (Figure 16–3). For patients who required low-dose warfarin (defined as <21 mg of warfarin per week), 35% of the dose estimations fell within 20% of the actual dose using the pharmacogenetic algorithm as compared with 24% using the clinical algorithm (P <.001) and 0% using the fixed-dose algorithm (P <.001). The pharmacogenetic algorithm also provided significantly less overestimations than the clinical and fixed-dose algorithms. For patients who required high-dose warfarin (defined as >49 mg of warfarin per week), 32.8% of the dose estimations fell within 20% of the actual dose using the pharmacogenetic algorithm as compared with 13.3% using the clinical algorithm (P <.001) and 0% using the fixed-dose algorithm (P <.001). The pharmacogenetic algorithm also provided significantly less underestimations than the clinical and fixed-dose algorithms. For patients who required intermediate-dose warfarin (defined as >21 and <49 mg of warfarin per week), there was no significant difference among the three approaches in estimating ideal warfarin doses. The study concluded that the pharmacogenetic dose algorithm that was developed provided better dose estimates than the clinical and fixed-dose algorithms for patients who required low or high doses of warfarin.
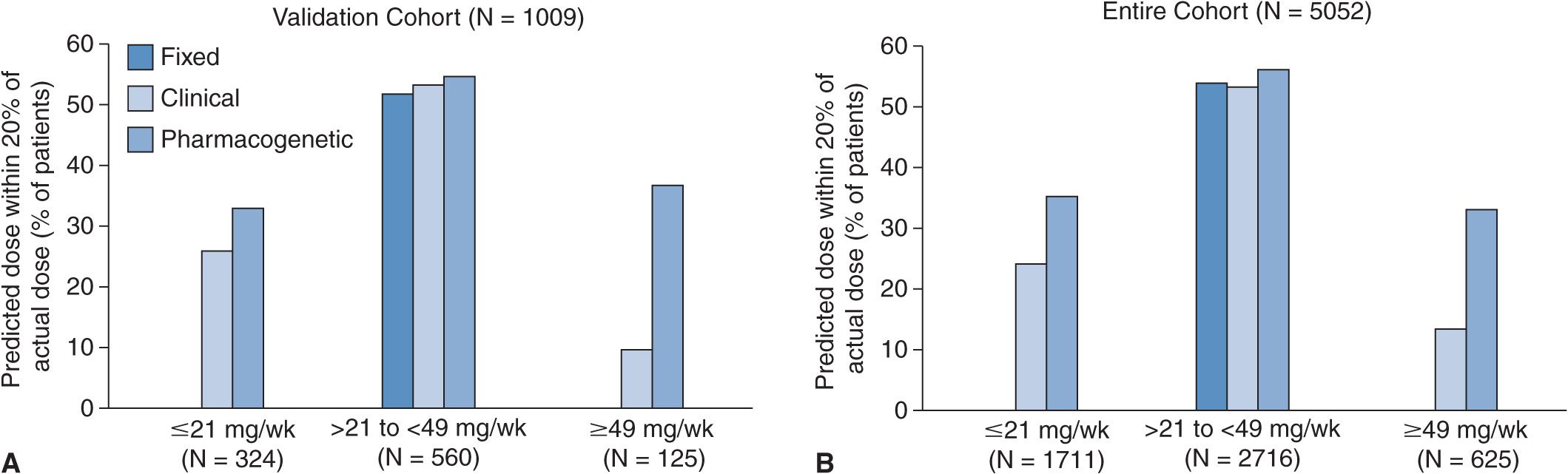
FIGURE 16–3 Warfarin pharmacogenetic dose algorithm.26
Another approved oral anticoagulant for nonvalvular atrial fibrillation is dabigatran etexilate, which is a prodrug that is rapidly hydrolyzed by plasma and hepatic esterases to dabigatran. Dabigatran further undergoes hepatic glucuronidation to active acylglucuronide isomers. The dosing for dabigatran etexi-late is fixed at 150 mg BID except for individuals with reduced renal function (i.e., CrCl 15–30 mL/min), in whom the dosing is 75 mg BID. Dabigatran is a direct thrombin inhibitor and, unlike warfarin, is not metabolized by CYP enzymes.27
The RELY trial compared two doses of dabigatran (110 mg BID and 150 mg BID) with adjusted-dose warfarin in the prevention of stroke in patients with atrial fibrillation.28 Results indicate that dabigatran 110 mg was noninferior to adjusted-dose warfarin in the prevention of thromboembolism and had less risks of bleeding (P <.001 and <.003, respectively). Dabigatran 150 mg was superior to adjusted-dose warfarin in the prevention of thromboembolism and had similar risks of bleeding (P <.001 and = 0.31, respectively).
Response to warfarin therapy is highly dependent on the CYP2C9 and VKORC1 genes. The CYP2C9 *2 and *3 variants are associated with decreased enzyme activity and decreased time to first therapeutic INR, increased rate of above 4.0 INRs, increased risk of bleeding, and lower maintenance doses. VKORC1 haplotype A is associated with faster time to therapeutic INR, faster time to an INR >4.0, and increased time over an INR >4.0, especially in the initial stages of therapy. The CYP2C9 and VKORC1 variants described require a lower dose of warfarin. This is especially important in the earlier stages of therapy, when a maintenance dose of warfarin has not yet been established. The risk of overanticoagulation is high. A patient’s response to warfarin may vary drastically depending on his or her genetic makeup.
ALZHEIMER’S DISEASE
AD is an age-related, progressive neurodegenerative disorder that develops over a period of years. The neurodegeneration is thought to begin 10–20 years before patients initially present with symptoms. The brain disorder begins with loss of memory and confusion and can later progress into behavior and personality changes, a decline in cognitive skills, and inability to make decisions and carry out activities of daily living (ADL). Age is the greatest risk factor for development of AD, but as more evidence emerges, genetics seem to play an increasingly important role. Currently, the disease affects one of eight patients over the age of 65, but 50% of patients over the age of 85.29 Other risk factors that are still under investigation for placing one at increased risk include female sex, lower levels of education, and history of head trauma. The hallmark signs of AD are amyloid plaques, neurofibrillary tangles (NFT), and loss of neurons responsible for memory and learning in the cortical areas and medial temporal lobe structures of the brain, which begin principally in the hippocampus and eventually affect all areas of the brain as neurodegeneration progresses.30
Two forms of AD exist: early onset Alzheimer’s disease (EOAD) and late-onset Alzheimer’s disease (LOAD). Individuals who develop symptoms before age 60–65 are considered to have EOAD that accounts for only 1–5% with dominantly inherited gene alterations on chromosomes 1, 14, and 21. LOAD has an onset after age 60–65 and is the predominant form of AD. Autosomal dominant AD occurs in at least three individuals over the span of two or more generations with two of the individuals as first-degree relatives. It represents less than 5% of cases and almost exclusively occurs in EOAD.31 Familial AD occurs in more than one individual; at least two of the affected individuals are third degree or closer. These are seen in LOAD (15–25%) but can also be seen in about 47% of EOAD. Sporadic AD occurs as an isolated case or cases are separated by more than three generations in a family, and it accounts for 7% of cases.31 Sporadic AD is usually LOAD, although there have been cases of EOAD (40%). LOAD is the predominant form of AD accounting for 95% of cases.
Amyloid Precursor Protein and Amyloid Plaques
Three deterministic autosomal dominant gene mutations have been implicated in the progression of amyloid plaques and EOAD: amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2). These mutations only account for 1–5% of EOAD cases and likely other genes are involved in EOAD.31,32 APP and PSEN1 demonstrate complete penetrance, whereas PSEN2 demonstrates 95% penetrance.31 Conversely APOE is a susceptibility gene for AD, with three isoforms ε2, ε3, and ε4. ε4 is associated with increased risk of AD, usually in LOAD, but also in EOAD. On the other hand, ε2 is thought to be protective.31 Most of the disease-associated genes differ by a single point mutation making them potential targets for gene silencing trials.32 Also because clinical trials are now focusing on manipulating the pathophysiology of the disease, genes affecting the development of amyloid plaques, NFT, and neurodegeneration will likely play a larger role in future treatments than in currently approved treatments.
APP is an integral membrane protein concentrated in synapses of neurons. Although its exact function is not known, it takes part in synapse formation, neural plasticity, and iron export.33 In normal conditions amyloid-beta is degraded by neprilysin, an insulin-degrading peptidase, and by endothelin-converting enzyme. It is then cleared from the brain by a balanced process mediated by low-density lipoprotein receptor-related protein and the receptor for advanced glycation end products. In AD, an imbalance between the production and clearance of amyloid-beta in the brain initiates the accumulation, ultimately leading to neuronal degeneration and dementia.30
Alterations in the protein or any component in the pathway can cause the buildup of A-beta 42 and lead to these senile plaques. Imbalances in the process begin with a mutation on chromosome 21 creating an abnormal APP.30 When there is a mutation on the APP protein, an abnormal A-beta is excised by the beta and gamma secretases, rather than being preferentially cleaved by alpha secretase. This form of A-beta cannot be cleared and catabolized because it does not complex with the exporter and is therefore built up over time into senile plaques. Thirty-two mutations have been identified as causing an amino acid substitution and cause conformational changes in the APP protein.34 Those affected by trisomy of chromosome 21 are more so affected because of an extra APP gene and tend to develop plaques earlier in life.35
A-beta is excised by two enzymes from APP labeled as beta-secretase and gamma secretase. Gamma secretase is an intramembranous protease complex consisting of four components: PSEN1, nicastrin, PSEN2, and APH-1. The PSEN proteins are thought to stabilize the gamma secretase.36 Beta-secretase activity comes from an integral membrane aspartyle protease called beta-site APP-cleaving enzyme (BACE-1).37 Mutations on both chromosomes 14 and 1 involve the formation of abnormal presenilin. When the components on the gamma secretase are mutated, the enzyme misprocesses APP, leading to an altered ratio between A-beta 42 and A-beta 40 and further deposition of A-beta 42 into plaques. With so many different mutations that are possible for PSEN1, the likelihood of developing AD in later stages in life is very high.38 It has been suggested that the gamma secretase may have substrates other than APP and the altered processing of these other substrates may also be involved in the pathophysiology of AD.39 Recent studies have shown that silencing the PSEN1 mutant gene is able to decrease the expression of the mutant PSEN1 protein and therefore a decrease in the accumulation of the toxic A-beta 42.32
APOE gene found on chromosome 19 may be involved in increasing risk. This gene codes for a protein that helps carry cholesterol in the bloodstream. APOE ε2 is the rarest form and may actually protect an individual against the disease. It may either protect a person or prolong the onset of the disease if present. APOE ε3 is the most common form and neither helps nor hurts the risk of developing Alzheimer. At least one of the APOE ε4 alleles occurs in 50–70% of the AD population. The risk increases in a dose-dependent manner where those with familial risk factors and more than one allele of the APOE e4 type will increase risk.31
Neurofibrillary Tangles
As AD progresses, NFT begin to spread throughout the brain starting in the neocortex. NFT are formed when tau, a micro-tubule-associated protein, becomes hyperphosphoyrlated. The tau-encoding gene, microtubule-associated protein tau (MAPT), consists of 16 exons.34 Tau assists in the formation and stabilization of microtubules.40 When tau is unable to bind to the micro-tubules, they become unstable and disintegrate. The aggregation of unbounded tau into an insoluble form results in pretangles that accumulate further into NFTs interfering with numerous cellular functions.30 A novel agent T-817MA, 1-(3-[2-(1-benzothiophen-5-yl)ethoxy]propyl)azeti-din-3-ol maleate, was studied in mice with the mutated tau gene and it seems to be a neuroprotective agent that can aid in inhibiting neuronal degeneration, thus improving motor and cognitive impairments.41
Fyn kinase is also thought to be involved in phosphorylating tau and exacerbating A-beta-mediated synaptic dysfunction. Fyn also increases the nonpathological cleavage of APP, suggesting that this opposes the progression of AD. In a study of transgenic mice, ultimately, loss of Fyn at early stages of the disease was found to increase soluble A-beta accumulation and worsens spatial learning in the absence of changes in tau phosphorylation.42
Other Genes
Researchers have begun using genome-wide associated studies (GWAS) to speed up the discovery process of risk factor genes. SORL1 gene on chromosome 11 was discovered as being another susceptibility gene involved in late-onset AD. It is involved in transporting APP within cells and when SORL1 was underexpressed, the beta amyloid levels increased and likewise when a similar gene, SORCS1, was underexpressed, gamma secretase activity and levels of amyloid increased.43
Another gene on chromosome 6, MTHFD1L, encodes methyltetrahydrofolate reductase (MTHFR) that converts folic acid to its active form, l-methylfolate, and is associated with risk of late-onset Alzheimer’s disease. Individuals possessing mutations may be twice as likely to develop the disease and may also possess elevated homocysteine levels in the body, which plays a role in AD.44 Therefore, in individuals with MTHFD1L mutations, therapy may focus on replacing the active form of folic acid, l-methylfolate theoretically (Deplin), but clinical trials need to be conducted to determine if this will be efficacious.
Current Treatments: Acetylcholinesterase Inhibitors and NMDA Antagonists
Current treatments for AD involve acetylcholinesterase inhibitors (AChEI) (donepezil, galantamine, rivastigmine) and moderate-affinity N-methyl-d-aspartate (NMDA) antagonist, memantine. In AD, there is a reduction in the activity of cholinergic neurons and AChEIs are employed to reduce the rate at which acetylcho-line (ACh) is broken down, thereby increasing the concentration of ACh in the brain and decreasing the loss of ACh caused by the death of cholinergic neurons.45,46 Memantine, on the other hand, is believed to protect neurons from excitotoxicity.34 Both of these approved pharmacological treatment options and most others in the pipeline only slow the apparent progression of AD; they do not improve cognitive decline, and they also do not modulate the genetic mutations. Once there are damages to the neurons and synapses, they cannot be reversed.
Response to AChEI can be altered by genetics. Donepezil and galantamine are metabolized by cytochrome P450 2D6, 3A4, and 1A2.47 CYP 2D6 genotypes consist of extensive metabolizers (EM), intermediate metabolizers (IM), PM, and UM and it is plausible that metabolizer status could affect efficacy or toxicity of cholinesterase inhibitors.48 Fifteen percent of Alzheimer patients may metabolize AChEI abnormally whereby about half would be UM, requiring higher doses. On the other hand, approximately half would be PM, increasing the likelihood of toxicity or adverse events.47 Patients with PM and UM phenotypes as well as ε4 genes tend to have worse treatment responses, whereas those with EM or IM phenotypes tend to respond more favorably.47 In addition, the gene encoding choline transferase (ChAT) that encodes the major catalytic enzyme of the cholinergic pathway is associated with response to AChEIs.49 ChAT is reduced in AD and is also correlated with the severity of dementia.49 Single nucleotide polymorphism (SNP) rs733722 in the promoter region of ChAT accounts for 6% of the variance in response to AChEIs.49 Another gene, SLC18A3, encodes for a vesicular ACh transporter that incorporates ChAT into synaptic vesicles and has also been implicated in disease susceptibility. rs733722 is present in approximately 33% of the population.49
Recent clinical trials have been aimed at interfering with A-beta deposition, vaccination with A-beta, selective A-beta 42-lowering agents, gamma secretase inhibition, and modulation of tau deposition. Unfortunately, many of the pitfalls of some of these trials have been significantly detrimental side effects or lack of significant improvement over placebo over extended periods of time.34 Although the currently approved treatments are only modestly affected by genetics, pharmacogenomics will continue to play an increasing role in Alzheimer’s treatments as newer treatments target the pathophysiology that is more tightly tied to genetics.
EPILEPSY
The estimated prevalence of epilepsy is 1%, resulting in over 50 million treated patients worldwide. Obtaining good seizure control without intolerable adverse effects can generally be achieved in 70–75% of patients; however, identifying the dose to achieve this is generally empiric. Unfortunately the introduction of several drugs newer than phenytoin (PHT), phenobarbital, and valproic acid has done little to improve seizure control in patients with refractory seizures. The understanding of genetic polymorphism in anticonvulsant metabolism enables less variability in dose concentration relationships. Pharmacogenomic factors are also useful in identifying patients with a higher susceptibility to adverse events unrelated to drug concentrations. Finally, pharmacogenomic factors have been studied to determine if anticonvulsant resistance leading to intractable seizures is due to overexpression of genes controlling energy-dependent efflux pumps leading to reduced exposure of the brain to that anticonvulsant.
Polymorphism in Anticonvulsant Metabolism
Of the current AEDs, old and new, the only drugs affected by cytochrome P450 polymorphisms are PHT and phenobarbital.50 While other AEDs are partly, or even extensively, metabolized by CYP enzymes, no evidence has been shown to correlate any CYP polymorphism with clinical response, serum drug concentrations, or other measurable outcomes.51 While both PHT and phenobarbital are metabolized by CYP2C9 and CYP2C19, more evidence exists that clinical outcomes rely on CYP polymorphisms with PHT than with phenobarbital. It has been shown that PHT metabolism varies based on various CYP2C9 alleles.52 This study demonstrated that the mean PHT dose required to achieve steady-state concentrations between 10 and 20 μg/mL in patients with one or two mutant alleles (CYP2C9*2 and CYP2C9*3) was 37% lower than that in patients homozygous for the wild type. In the same study, the PHT doses required to achieve therapeutic steady state were not significantly different between carriers of the CYP2C19 mutant allele. Case studies have also reported unexpected PHT toxicity in patients with at least one mutant CYP2C9*3 allele. One such case study reported a 41-year-old woman who on admission to the hospital for seizures was given 15 mg/kg over 30 minutes and became nonresponsive with low respiratory rate.53 Subsequent serum PHT concentration was 79 μg/mL and genotyping revealed she carried one wild-type and one mutant CYP2C9*3 allele.
A study of Japanese epileptics using phenobarbital showed a correlation to genotype and clearance of phenobarbital.54 This study measured serum concentrations and calculated clearance rates for 79 patients treated with phenobarbital and genotyped them for both CYP2C9 and CYP2C19. The authors found that patients with a mutant CYP2C9 allele had significantly reduced clearance of phenobarbital compared with wild-type homozygotes. They found no correlation between CYP2C19 genotype and phenobarbital clearance.
This suggests that there may be utility in genotyping for CYP2C9 to determine patients who may have reduced metabolism of PHT or phenobarbital before initiating therapy with these agents. These results also suggest that although both drugs are apparent substrates of CYP2C19, the genotype of the patient has not been shown to correlate to clinical outcomes such as maintenance dose or clearance.
Genomic Predictors of Higher Susceptibility to Adverse Events Unrelated To Drug Concentrations
Drug hypersensitivity reactions that commonly involve skin and mucosa can, in severe cases, result in aplastic anemia, renal failure, hepatitis, and death. Carbamazepine, an older but effective anticonvulsant, is also used widely for mood stabilization in bipolar disorder, for neuropathic pain syndromes, and an adjunct in chronic pain syndromes. In rare cases it can cause the life-threatening hypersensitivity reactions of Stevens–Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN). While both are rare, they are clearly associated with AEDs but more commonly seen with carbamazepine.55 Recent research has shown that the human leukocyte antigen (HLA) HLA-B*1502 shows a strong association with carbamazepine-induced SJS in Han Chinese.56 Although this association was not found in any Caucasian populations, the finding prompted the FDA to change the labeling information of carbamazepine to include information about HLA-B*1502, and the FDA now recommends genotyping individuals of Asian ancestry for the allele prior to using the drug.57 After the FDA recommendations were published, another study suggested a strong association between the allele and both carbamazepine- and PHT-induced SJS in a Thai population, providing evidence that the correlation may not be limited to Han Chinese.58 Considering the structural similarities between carbamazepine and other AEDs, a study was conducted by Hung et al. to test whether a similar association existed between the HLA-B*1502 allele and drug-induced SJS/TEN during therapy with other commonly used AEDs, specifically PHT, lamotrigine (LTG), and oxcarbazepine (OXC).59
This was a case–control association study by identifying cases of SJS and TEN induced from PHT, LTG, or OXC from various hospital systems in Taiwan from 2002 to 2008. The authors enrolled 26 PHT-induced cases, 6 LTG-induced cases, and 3 OXC-induced cases. In all enrolled cases, the drug was considered the cause of the SJS/TEN if onset was within 2 months of starting the drug. For control cases, 113 patients using PHT and 67 using LTG for over 3 months without any adverse drug reactions were enrolled. To control for OXC, a Taiwanese population study was used and 93 healthy subjects were randomly selected. All enrolled individuals were Han Chinese living in Taiwan. Individuals were then HLA genotyped to determine A, B, C, and DRB1 alleles. The authors found HLA-B*1502 present in 30.8% of PHT-related SJS/TEN, 33% of LTG-related SJS, and 100% of OXC-related SJS with odds ratios of 5.1 (95% CI: 1.8–15.1; ![]() ), 5.1 (95% CI: 0.8–33.8;
), 5.1 (95% CI: 0.8–33.8; ![]() ), and 80.7 (95% CI: 3.8–1,714.4;
), and 80.7 (95% CI: 3.8–1,714.4; ![]() ), respectively. HLA-B*1502 is a common risk allele for multiple aromatic anti-epileptic drugs.
), respectively. HLA-B*1502 is a common risk allele for multiple aromatic anti-epileptic drugs.
Considering the potential for SJS or TEN, the FDA recommends genotyping patients of Asian ancestry prior to treatment with carbamazepine. Combined with evidence that other AEDs may have the same increased risk with the HLA-B*1502 allele, genetic testing for Asian patients may prove extremely useful for reducing the risk of this adverse reaction. The association between the allele and carbamazepine has not been demonstrated in non-Asian populations.60 This is likely because SJS/TEN and the frequency of the allele in non-Asians are so rare that the sample sizes are not large enough to detect it.
Pharmacogenomic Factors and Anticonvulsant Resistance
It has been hypothesized that patients with uncontrolled seizures despite treatment with multiple anticonvulsants may have a genetic overexpression of energy-dependent drug efflux transporters at the blood brain barrier (BBB) rendering them anti-convulsant resistant. Several studies have investigated whether polymorphisms in ABCB1—the gene that encodes the efflux transporter ABCB1 (also known as P-glycoprotein [P-gp] and MDR1)—are correlated with the nonresponse to antiepileptic drugs, with conflicting results.
In a report by Siddiqui et al., 115 drug-responsive epileptics, 200 drug-resistant epileptics, and 200 control patients were genotyped at ABCB1 3435 for the C or T polymorphism.61 There was a significantly higher likelihood for drug-resistant epileptics to have the CC genotype rather than the TT genotype compared with drug-responsive epileptics with an odds ratio of 2.66 and a 95% confidence interval of 1.32–5.38. Unfortunately these results could not be replicated by two subsequent and similar studies.62,63
While a genetic variation of the ABCB1 transporter is a reasonable potential cause of multiple drug resistance in epileptics, it does not seem that response or nonresponse can be predicted from the C3435T polymorphism. It would seem possible that either anticonvulsants are not dependent on P-gp for transport into the brain or anticonvulsants are not high-affinity substrates for P-gp. That P-gp is not involved in drug resistance to anticonvulsants can also be seen in animal data. Data shown in Figure 16–1 show that there is little, if any, significant change in brain/plasma ratio following a known P-gp inhibitor for several anti-convulsants.50 Moreover, the relationship between plasma and CSF concentrations for older anticonvulsants is linear rather than nonlinear as would be expected if an energy-dependent transporter were involved. These results do not necessarily suggest that variation in the ABCB1 transporter plays no role in multiple drug resistance, but instead suggest that whatever role the transporter plays in resistance may not be clearly predicted by genotyping for the C3435T polymorphism. Conflicting results may be further explained by Kimchi-Sarfaty et al. These authors explain that a synonymous SNP found in the ABCB1 transporter gene results in an altered P-gp response, despite producing no changes in the amino acid that is produced. The authors hypothesize that, although the amino acid composition itself remains unchanged, this rare codon causes altered protein folding and therefore conformational changes at the P-gp receptor, and, thus, can change the substrate-receptor response.64 Clearly, more research should be performed to determine if these changes translate into clinically meaningful effects on drug response.
In summary, genomic study is clearly useful in identifying populations of patients likely to need higher or lower doses of PHT and phenobarbital owing to differences in rate of metabolism. Genomic study is also useful in identifying populations of patients and individual patients more susceptible to hypersensitivity reactions. Unfortunately, existing data do not suggest that genomic study will be useful in explaining anticonvulsant resistance.
MULTIPLE SCLEROSIS
MS is a chronic autoimmune disease of the central nervous system involving pathological inflammation, demyelination, and neuron degeneration characterized by progressive physical and cognitive dysfunction.65 The pathogenesis of MS is not fully understood, but appears to involve both environmental and genetic mechanisms. The clinical course of MS is extensively variable between individuals. An initial triggering event is thought to produce a subclinical presentation of neurodegenerative changes. This latent period is succeeded by an initial demyelinating event, called a clinically isolated syndrome (CIS). MS diagnosis is confirmed on a second isolated event and evidence of new inflammatory lesions on brain MRI. MS may remain subclinical in some individuals throughout an entire lifetime, while in others, an initial clinical isolated syndrome is followed by a mild course of disease. Eighty-five percent of patients develop a relapsing-remitting course (RRMS), which can progress into secondary progressive MS (SPMS) characterized by progressive neurological decline without periods of remission. In 10–15% of cases, disease is progressive from the initial event and is termed primary progressive (PPMS) that may also be relapsing in nature (PRMS).
The susceptibility of individuals to developing MS appears to be heterogeneous and involves complex non-Mendelian inheritance.66,67 Risk factors for developing MS appear to include female sex, family history of MS, and Caucasian ethnicity.67,68 Environmental factors have also been explored. Antibodies against the Epstein–Barr virus have been associated with gray matter atrophy in MS.69,70 Smoking is associated with greater disability, lesions, and neurodegeneration in MS.71,72 Additionally, vitamin D deficiency and polymorphisms in the vitamin D receptor have been associated with increased risk of developing MS and accelerated disease progression.73 The improvement of genetic testing, particularly of genome-wide association studies (GWAS), has identified several genes that may confer susceptibility to developing MS. In particular, the polymorphisms in the HLA have consistently shown to confer 20–60% of the genetic susceptibility to MS. Polymorphisms in other immune system components, including IL2RA, IL7RA, CD58, CLEC16A, and CD226, may increase disease susceptibility.66,74
Given the extensive heterogeneity of susceptibility and clinical course of MS, treatment of MS is equally complex. Current strategies aim at symptomatic management and delaying disease progression, but do not offer a cure. Disease-modifying therapies downregulate various pathogenic immune processes and have shown to effectively reduce relapses, lessen brain MRI activity, and slow progression of disability.75,76 Despite these advantages, studies indicate large differences in treatment response between individuals. While some patients appear to respond well, up to half of those treated are nonresponders to therapy, and some develop serious side effects.77,78 Interestingly, treatment failure with one agent does not appear to correlate with ineffectiveness of a second agent. The ability to predict patient response would allow for individualized therapy that may reduce both the economic and disease burdens of MS. Pharmacogenomic research has attempted to identify genetic differences that may provide insight into this puzzle. The ability to predict patient response would allow for individualized therapy that may reduce both the economic and disease burdens of MS.
Interferon-β
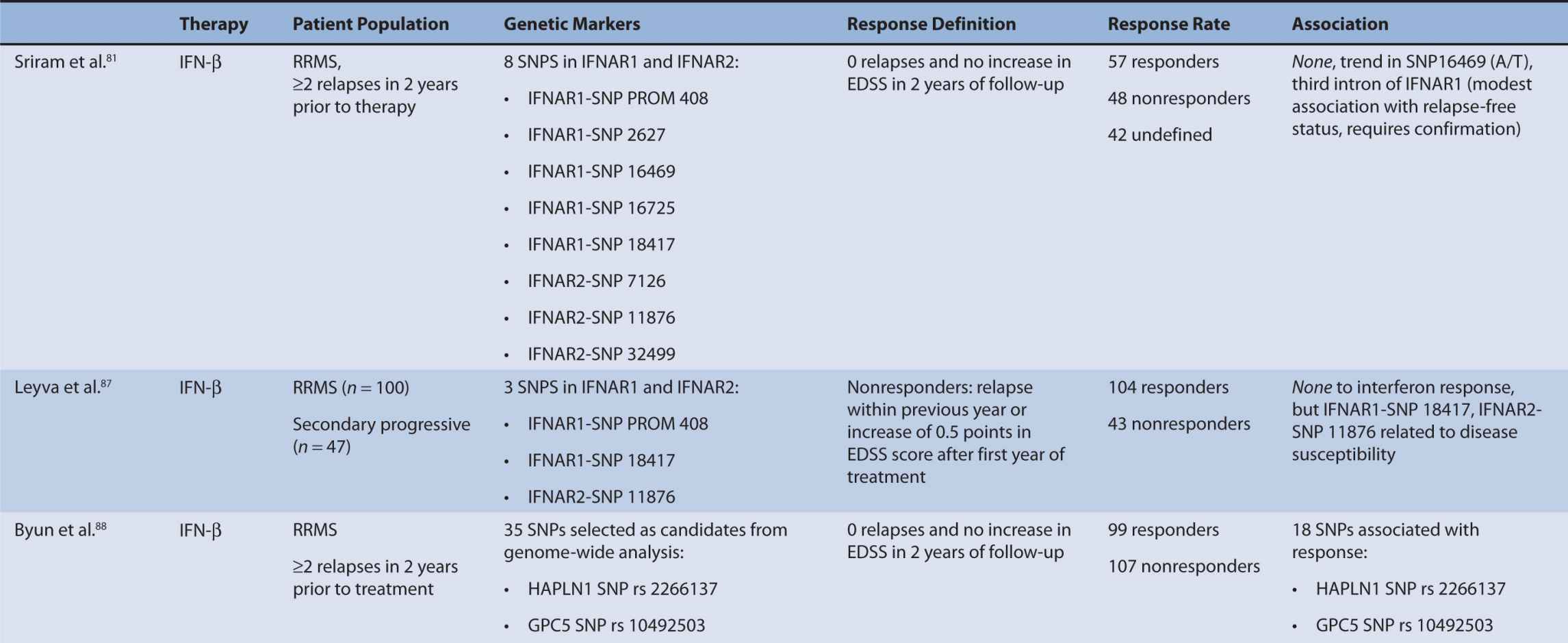
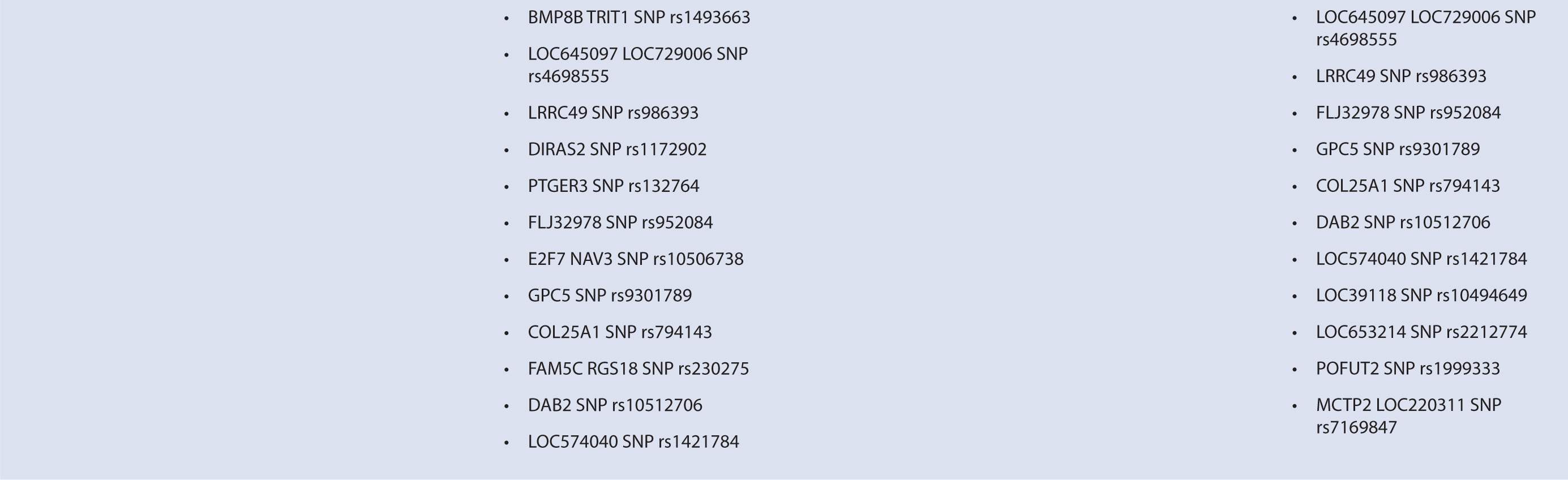
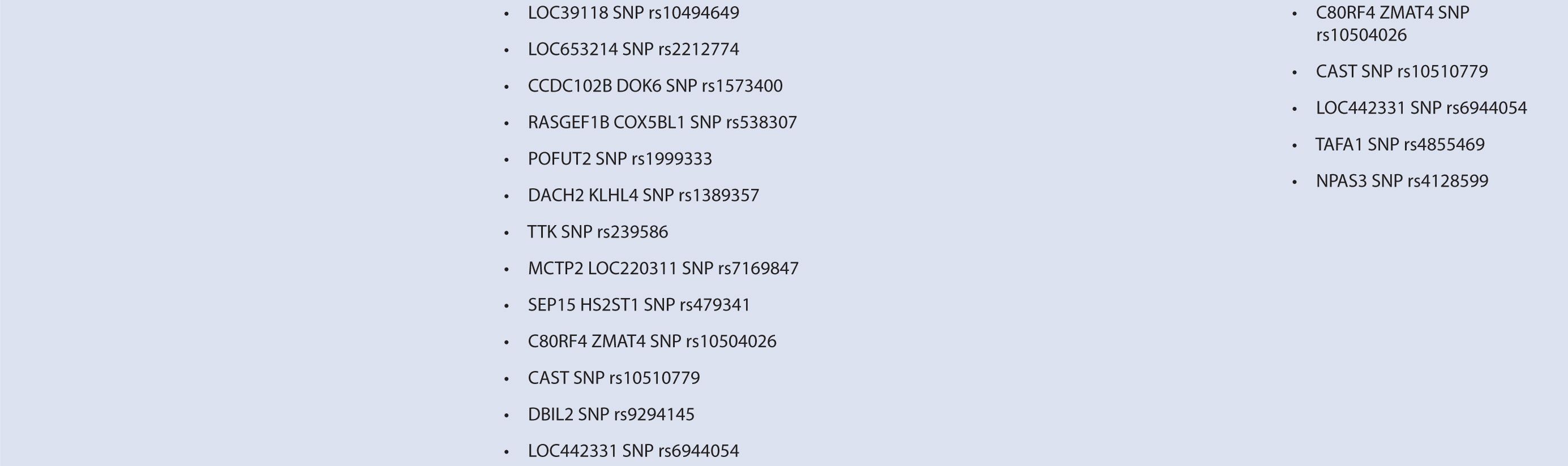
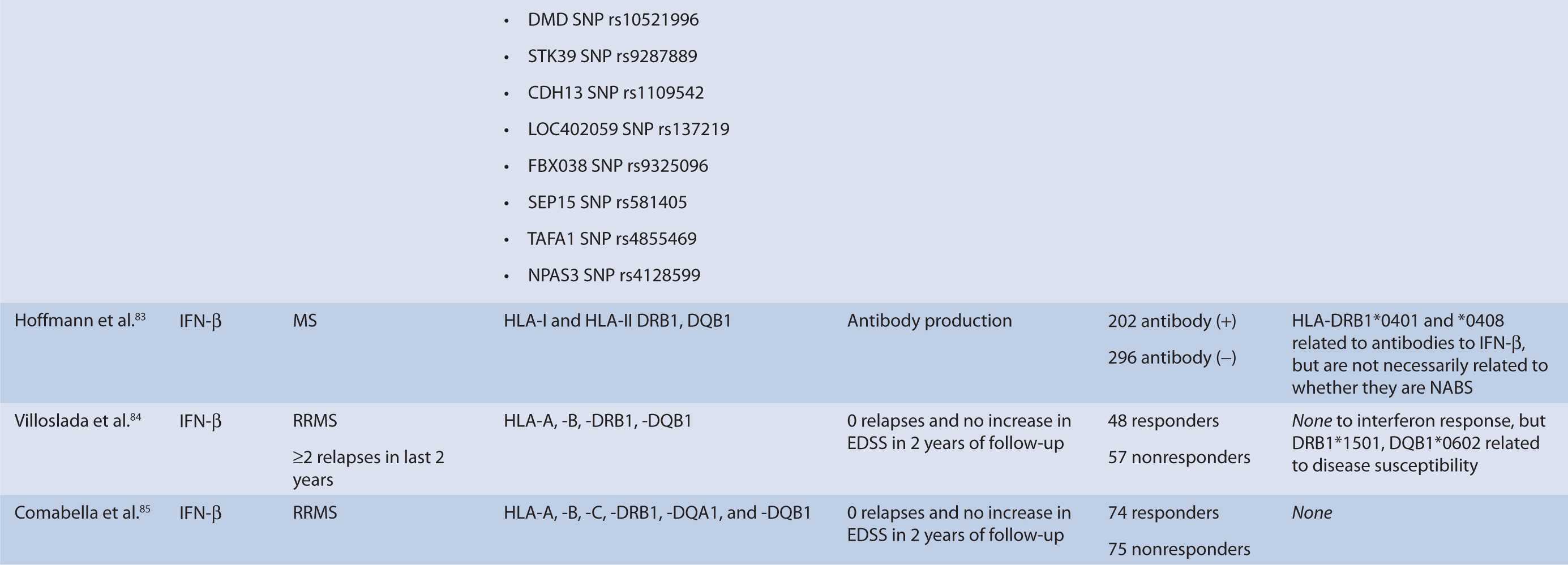
Interferon therapy has been used as a first-line treatment for MS for many years. Interferon-beta is a naturally occurring immunoregulator in humans and has antiproliferative, proapoptotic, and antiviral properties.79 The precise mechanism of action of IFN-β in MS is unknown; however, it may involve inhibition of production of proinflammatory cytokines, decreased brain penetration of T-cells, and promotion of remyelination.80 There are currently three interferons approved for use in MS, including Avonex (IFN-β1a), Rebif (IFN-β1a), and Betaferon (IFN-β1b). All of these interferons bind to the same receptor, IFNAR, which consists of two subunits, IFNAR1 and IFNAR2. These receptors are coded by genes on chromosome 21q22.1, which are under study.81 Side effects from interferon therapy include injection site reactions, flu-like symptoms, depression, bronchospasms, and hepatotoxicity. In responders, interferon therapy decreases relapse rate and severity and slows clinical progression; however, relapse rates occur in up to 50% of people treated with interferon.82 IFN-β therapy has been shown to evoke antibody production in some individuals. These antibodies can lead to immunogenic responses and can neutralize IFN-β and directly decrease therapeutic efficacy. The development of binding antibodies (BABs), some of which are neutralizing (NABs), are thought to decrease the efficacy of interferons to varying degrees.83 Given the incongruity in treatment response to interferon therapy, genomic studies have attempted to identify loci that may predict treatment response among MS patients. In particular, candidate gene studies were performed for HLA and interferon receptor polymorphisms, and one genome-wide analysis was performed. Studies varied greatly in response definition and genetic marker studies (Table 16–1).
TABLE 16–1 Pharmacogenomic studies of disease-modifying therapies in multiple sclerosis.


Human Leukocyte Antigen
HLA are critical components of T-cell activation and differentiation, and HLA alleles have consistently shown to confer disease susceptibility in MS and may be involved in disease progression. They are located on major histocompatibility complex (MHC) on chromosome 6p21.3.84 Therefore, HLA alleles have been studied extensively in relation to treatment response of disease-modifying therapies and association with antibody production against IFN-β. HLA-A, -B, -C, -DRB1*1501, and -DQB1*0602 have been studied. No association was found with HLA status and treatment outcome, but DRB1*1501 and DQB1*0602 were related to disease susceptibility.85,86
Hoffmann et al.83 studied the association of HLA class II alleles and the development of and treatment response in MS patients in Germany. Patients carrying the HLA-DRB1*0401 and -DRB1*0408 carriers had increased risk of developing IFN-β antibodies. However, these alleles were not consistently associated with NAB production. Although allelic variation has been correlated with antibody production, it has not been directly correlated with treatment response.
Interferon Receptors
The primary mechanism of interferon action is thought to involve binding of interferon to the interferon type I receptor, a cell-surface receptor responsible for activation of the JAK-STAT pathway and subsequent stimulation of an immunological signaling cascade.86 The interferon receptor is comprised of two subunits, IFNAR1 and IFNAR2, which have been tested for polymorphisms in association with treatment response. These receptors are coded for by genes on chromosome 21q22.1, which are under study.81 Only SNP 16469 on the IFNAR1 gene showed a moderate association with relapse-free status.81 However, IFNAR1 (SNP 18417) and IFNAR2 (SNP 11876) have shown a relationship to disease susceptibility.87 Conversely, Byun et al. did not find a relationship between SNPs detected in or near IFNAR1 or IFNAR2, but the findings were limited as the authors analyzed only SNPS detected by their microarrays.88
Genome-Wide Analysis
Byun et al.88 performed a genome-wide analysis to assess the role of various polymorphisms in interferon treatment response. Two hundred and six patients were recruited at four medical centers in the Mediterranean Basin. Patients needed to have RRMS, current treatment with beta-interferon, and a history of two or more relapses in 2 years prior to therapy initiation for inclusion. They were followed for 2 years with disease assessments by neurologists every 3 months. Clinical response was defined as zero relapses and no increase in EDSS throughout the entire 2-year follow-up period. Overall, 99 patients were considered responders while 107 were nonresponders. Genome-wide analysis of patient DNA was used to identify potential SNP candidates based on allelic frequencies and clustering. Thirty-five candidate SNPs were selected, 18 of which remained significant after analysis, including glypican, collagen type XXV α1, hyaluronan proteoglycan link protein, calpastatin, TAFA1 (chemokine-like), LOC442331 (similar to dynein), and neuronal PAS dominant protein 3 which are located within genes. The remaining SNPS are in intergenic regions. Of note, SNPs related to ion channels and signal transduction pathways varied most frequently among responders and nonresponders. In particular, glypican 5 SNPs were common. Glypicans alter cytokine responsiveness and aid in axon regeneration and synapse formation, and are associated with MS plaques. Peripherally, they are also required for Schwann cell myelination. Interferon therapy may alter expression of glypicans and SNPs could potentially alter this effect. Polymorphisms in hyaluronan proteoglycans and collagen may affect the binding of matrix metalloproteases released by leukocytes to aid in their penetration through basement membranes. Interferon-beta has shown to inhibit these proteins. SNP variations in these proteins may prevent IFN-β’s inhibitory effect. Interestingly, no association was found with interferon receptors, further corroborating reports of previous trials (Table 16–1). This study was limited in that it could only examine SNPs detected by the authors’ microarray and that the authors did not control for NABs or BABs.88
Glatiramer Acetate (Copaxone)
Glatiramer acetate (GA) is a peptide similar in structure to myelin basic protein, which may modify the immune response of myelin antigens in MS patients and increase repair of neural tissue.89 In clinical trials, Copaxone produced a 30% reduction in relapse rate, increased time between relapses and number of patients who were relapse-free, and did slow disease progression in MS patients. Injection site reactions are common and use of GA may produce transient chest pain, flushing, and palpitations. Two studies to date have looked at the role of genetic markers in conferring treatment response in patients receiving GA therapy. GA binds to MHC class II molecules, some of which are encoded by DRB1.
Gross et al.90 performed a retrospective analysis on the association with the HLA-DRB1*1501 allele as well as interferon response factor 8 (IRF8) in time to first event in patients treated with either IFN-β or GA (Table 16–1). Time to first event was defined as clinical relapse, change in T2 hyperintense lesion or new gadolinium-enhancing lesion on MRI, or an EDSS increase of 1 or more points over 6 months. Overall, time to first event was not different between GA and IFN-β. The HLA-DRB1*1501 allele correlated with a better treatment response in GA-treated patients. Association of this allele was not found in the IFN-β subset. Although this association explained some of the hazard-free survival in GA-treated patients, the authors conclude that it likely only exerts a modest effect on treatment response with GA therapy. In addition, those homozygous for IRF8 had demonstrated longer times to first event in the GA population more than the IFN-β population and appeared to be associated with shorter time to first event in the IFN-β population.90 However, this was not able to be replicated in IFN-β treated subjects with negative antibody status. IRF8 is associated with susceptibility to MS and upregulation of interferon response genes and patients with increased interferon response genes seem to be at an increased risk of relapse.90
Grossman et al. recruited patients from 2 previous clinical trials of GA versus placebo treatment, and assessed the relationship between treatment response and 63 SNPs.91 One hundred and one patients from the European/Canadian MRI trial and 73 patients from the US pivotal trail were enrolled and genotypes were analyzed. Responders in the E/C trial were previously defined as having no clinical relapse in the 9 months of follow-up and no more than one T1-enhancing lesion, or no new T1-enhancing lesion appearance in the first 6 months of therapy, while US trial responders were identified as patients who remained relapse-free with no increase in EDSS score over the duration of the 2-year trial. Results indicated two genes were predictive of GA response, TRB@ and CTSS. TRB@ is a molecular complex thought to be associated with GA mechanism of action, and was associated with treatment response in both E/C and US responders (OR 6.85). CTSS may be involved in GA metabolism, and was associated with treatment response in the E/C trial. In addition, it plays a role in MHC II chain processing. Of note, the HLA-DRB1*1501 was not associated with drug response, but the study may have been underpowered to detect an association. Nominal significance was seen for MBP, CD86, FAS, IL1R1, and IL12RB2. MBP is thought to be the primary myelin autoantigen attacked by the immune system, which is inhibited by GA.
Natalizumab (Tysabri)
Natalizumab is a recombinant humanized monoclonal antibody that inhibits VLA-4 adhesion to VCAM expressed on activated endothelium.92 Inhibition of VLA-4 results in impaired T-cell migration across the BBB and apoptosis. In patients with RRMS, natalizumab reduces the probability of disease progression, increases probability of remaining relapse-free, and reduces the appearance of new lesions on MRI. Despite treatment response in most patients, Tysabri is currently considered a second-line agent in the treatment of MS due to the risk of progressive multifocal leukoencephalopathy (PML).93 PML is caused by JC polyomavirus (JCV), an opportunistic pathogen which infects oligodendrocytes and causes rapid neuronal demyelination and death. Although an estimated 50–60% of MS patients are infected with JCV, most individuals will remain asymptomatic unless immunocompromised. JCV exists in two distinct subtypes, a benign form and a pathogenic form capable of infecting nerve cells. This pathogenic form has been associated with multiple mutations of the noncoding control region (NCCR) involved in gene replication.
To identify how JCV genotype or specific mutations may confer susceptibility to developing PML, Reid et al.93 conducted a genomic analysis of viral isolates collected from 17 PML patients treated with natalizumab. Isolates were obtained from plasma, urine, and CSF of selected patients and JCV genotype, NCCR variations, and VP1 capsid protein sequences were analyzed using PCR amplification. Results indicated multiple JCV genotypes between individuals, with each patient being infected with a single genotype. No association was found between JCV genotype and PML development. In blood and CSF, NCCR and VP1 mutations were found in 100% and 81% of isolates, respectively. In contrast, urine samples of JCV contained no mutations in these regions. These results indicate that after infection with the benign form of the virus, NCCR and VP1 mutations occur via an unknown mechanism that creates the pathogenic, infectious form of the virus responsible for PML. How natalizumab may act to facilitate this transformation is unknown, but warrants further study. Identification of susceptibility loci on the viral genome may help identify patients at risk for PML development and allow for more selective use of natalizumab.
Fingolimod (Gilenya)
Fingolimod is the first oral therapy available for MS and reduces frequency of relapses and delays disability.94 It binds to sphingosine 1-phosphate receptor subtypes 1, 3, 4, and 5, which reduces lymphocyte levels in the blood by inhibiting release from lymph nodes. In a clinical trial comparing fingolimod with interferon-β, patients experienced lower annual relapse rates (17% vs. 30%) and reduced appearance of gadolium-enhanced lesions on MRI.95 Side effects of fingolimod include macular edema, transient bradycardia on initial administration, and immunosuppression. At present, no pharmacogenomic studies have been performed on fingolimod, as heterogeneous treatment responses or side effect profiles are not yet apparent with this therapy.
Mitoxantrone (Novantrone)
Mitoxantrone (MX) is an antineoplastic agent indicated for SPMS and has shown to reduce relapse rates and decrease the rate of disability.96 MX inhibits DNA and RNA synthesis and has many immunomodulatory effects that are thought to influence MS disease progression. Currently, the clinical utility of MX is limited due to an increased risk of hematological malignancy and dose-limiting cardiotoxicity.
Therapeutic efficacy of MX appears to be influenced by polymorphisms in the ATP-binding cassette transporter genes responsible for drug efflux. Cotte et al. studied the association between ABC polymorphisms and treatment response and side effect profile of MS patients treated with MX.97 Genotypic data were analyzed retrospectively from 309 MX or MX-corticosteroid-treated patients for ABCB1 ![]() ,
, ![]() , ABCG2 V12M, and Q141K. Clinical response was analyzed between 9 and 12 months, and defined by EDSS stability or improvement, improvement in relapse rate, MSFC stability/improvement, MRI activity, and physician discretion. In patients receiving MX monotherapy, 78.1% were considered responders and 59.1% of MX/CS-treated patients were considered responders. Twenty-eight patients experienced severe cardiac effects and eight had hematological effects. When compared with healthy subjects, no difference was found in genotypic frequencies. An in vitro assay was performed to assess ABC-transporter-mediated MX efflux in CD56+ cells. Patients homozygous for common ABCB1/ABCB2 alleles had higher MX efflux and less CD56+ cell death than patients with at least one variant allele. When correlated with therapeutic response, patients with two variant alleles had higher response rates (83%) than patients with two common alleles (62.5). Heterozygous patients had an intermediate effect (79.6%). In MX/CS-treated patients, no association was found between variant ABC alleles and therapeutic response; however, this cohort was small. No correlation was found between ABC-transporter alleles and cardiotoxicity or hematotoxicity. It appears ABCB1 and ABCG2 alleles may be useful in identifying potential responders to MX treatment.
, ABCG2 V12M, and Q141K. Clinical response was analyzed between 9 and 12 months, and defined by EDSS stability or improvement, improvement in relapse rate, MSFC stability/improvement, MRI activity, and physician discretion. In patients receiving MX monotherapy, 78.1% were considered responders and 59.1% of MX/CS-treated patients were considered responders. Twenty-eight patients experienced severe cardiac effects and eight had hematological effects. When compared with healthy subjects, no difference was found in genotypic frequencies. An in vitro assay was performed to assess ABC-transporter-mediated MX efflux in CD56+ cells. Patients homozygous for common ABCB1/ABCB2 alleles had higher MX efflux and less CD56+ cell death than patients with at least one variant allele. When correlated with therapeutic response, patients with two variant alleles had higher response rates (83%) than patients with two common alleles (62.5). Heterozygous patients had an intermediate effect (79.6%). In MX/CS-treated patients, no association was found between variant ABC alleles and therapeutic response; however, this cohort was small. No correlation was found between ABC-transporter alleles and cardiotoxicity or hematotoxicity. It appears ABCB1 and ABCG2 alleles may be useful in identifying potential responders to MX treatment.
Conclusions
The current body of evidence indicates that pharmacogenomic analysis may play an important role in both the efficacy and toxicity of MS treatments. In patients treated with beta-interferons, polymorphisms in the interferon receptor or HLA do not correlate with treatment response, while HLA-DRB1*0401 and *0408 loci correlate with antibody production. Recently, genome-wide analysis revealed SNPs in glypticans and extracellular matrix proteins show a relationship to treatment response. Increased time to first event with GA therapy has been associated with HLA-DRB1*1501, and TRB@ and CTSS genes were associated with treatment response. PML found in natalizumab patients may be associated with specific viral polymorphisms of the JCV. MX responsiveness appears to be related to ABCB1/ABCB2 polymorphisms.
The aforementioned pharmacogenomic studies have been useful in identifying possible mechanisms conferring response or toxicity to treatment. The continued utilization of genome-wide screening will expectantly provide more information on the complex genetic and environmental interactions involved in MS susceptibility, disease course, and treatment. These findings will be crucial in improving the clinical course and outlook of MS.
PARKINSON DISEASE
PD is a degenerative neurological disorder that results in death of dopaminergic neurons in the substantia nigra. The exact etiology of PD is yet to be determined, but it has been hypothesized that both genetic and environmental factors may play a role. The loss of dopaminergic neurons of the nigrostriatal pathway results in inhibition of thalamic activity, which thereby reduces activity of the motor cortex. This decrease in motor function results in the hallmark features of PD including tremor at rest, bradykinesia, postural instability, and rigidity. Advanced stages of the disease may even precipitate behavioral and cognitive impairment.98
Although there is no cure for PD, several pharmacological treatments have proven to be efficacious in reducing motor symptoms and improving quality of life. Such classes of medications include dopamine precursors, monoamine oxidase type B (MAO-B) inhibitors, catechol-O-methyltransferase (COMT) inhibitors, dopamine receptor agonists, and NMDA receptor antagonists. However, the interindividual variability with respect to both treatment response and tolerability is large.
In recent years, there has been growing interest in genetic variability as an explanation for the large interindividual variability in antiparkinson drug efficacy and toxicity. Numerous gene candidates have been identified including, but not limited to, genes encoding COMT (COMT gene), dopamine receptors (DRD1, DRD2, DRD3 genes), dopamine beta-hydroxylase (DBH gene), dopamine transporter (DAT gene), monoamine oxidases (MAOA, MAOB genes), and cytochrome P450 enzymes.99
Dopamine Precursors
Levodopa (L-dopa) is the immediate metabolic precursor of dopamine and is the cornerstone treatment for PD. L-dopa is administered with carbidopa, a noncompetitive l-amino acid decarboxylase inhibitor that prevents the conversion of L-dopa to dopamine in the periphery and GI tract. Since carbidopa does not cross the BBB while L-dopa does, concomitant administration of these two drugs substantially increases the bioavailability of L-dopa in the central nervous system. Once past the BBB, L-dopa is converted to dopamine by aromatic l-amino acid decarboxylase (DOPA decarboxylase) in the substantia nigra where it is stored in presynaptic dopaminergic neurons. When stimulated for release, the decarboxylated L-dopa reserve binds to postsynaptic D1 and D2 receptors.
Dopamine and its precursors undergo immediate degradation by the enzymes COMT and MAO-B.98 Genetic polymorphisms of COMT and MAO-B enzymes have been hypothesized to influence the effectiveness of L-dopa therapy. Perhaps the most studied genetic polymorphism of the COMT enzyme is the Val158 Met polymorphism, which is an SNP caused by a valine to methionine substitution. This SNP results in a low-activity allele COMT L and may result in three possible genotypes with varying activity—low-activity COMTL/L, intermediate-activity COMTL/H, and high-activity COMTH/H. The COMTH/H variant is up to four times more active in the degradation of dopa-mine versus the low-activity variant.
Reilly et al. were one of the first to correlate COMT activity with patient response to L-dopa therapy.100 Their study in 14 patients found that those with higher erythrocyte COMT activity experienced less favorable clinical responses to L-dopa than patients with lower erythrocyte COMT activity. Their study set the stage for future research into the genetic polymorphisms that affect the activity level of the COMT enzyme. Bialecka et al. demonstrated a possible link between SNPs of the COMT gene and interpatient variability in therapeutic response as measured by the daily optimal L-dopa dose during the fifth year of treatment.101 This case–control study of 679 patients of Caucasian origin observed that the mean L-dopa dose at year 5 of treatment increased with the activity of functional haplotypes. Significantly higher mean doses (604 mg per day vs. 512 mg per day, P <.05) were associated with high-activity COMTH/H haplotype carriers as compared with low-activity COMTL/L haplotype carriers. However, COMT genotype was not found to be associated with the development of L-dopa-induced dyskinesias. Yet, a smaller retrospective study conducted by Bialecka et al. failed to observe significant differences between COMT and MAO-B genotypes on effective L-dopa daily doses.102
In contrast, a prospective study of 104 PD patients of Italian descent conducted by Contin et al. investigated the effect of COMT Val158 Met polymorphism on L-dopa response and rate of adverse events using pharmacokinetic (Tmax) and pharmacodynamic (Cmax) parameters.103 Neither did they find statistically significant differences in pharmacokinetic and pharmacodynamic parameters in patients with varying COMT haplotypes nor were they able to correlate the COMT polymorphisms with oral bioavailability in their study population. Similar studies in Korean and Japanese patients also found no statistically significant differences between COMT genotypes and pharmacokinetic and pharmacodynamic responses to L-dopa therapy.104,105
Associations between genetic polymorphisms and L-dopa-induced side effects, particularly dyskinesias, have also been examined to some extent. Several investigators observed statistically significant correlations between L-dopa-induced peak-dose dyskinesias with the DRD2 CAn-STR gene polymorphism, which include four common alleles (13, 14, 15, or 16 CA repeats). Oliveri et al. found that patients with the at least 1 of the 13 or 14 alleles of the DRD2 CAn-STR gene had a risk reduction in development of dyskinesias when compared with those who carried none (![]() , 95% CI: 0.11–0.77).106 Zappia et al. confirmed their result only in male subjects (
, 95% CI: 0.11–0.77).106 Zappia et al. confirmed their result only in male subjects (![]() , 95% CI: 0.14–0.84).107 Strong et al. observed that patients with the 14 allele or 14/15 genotype of the DRD2 CAn-STR gene actually had an increased risk of early dyskinesia (
, 95% CI: 0.14–0.84).107 Strong et al. observed that patients with the 14 allele or 14/15 genotype of the DRD2 CAn-STR gene actually had an increased risk of early dyskinesia (![]() , CI: 1.1–10.4;
, CI: 1.1–10.4; ![]() , CI: 1.4–510).108
, CI: 1.4–510).108
As evidenced by the mixed results in the studies just previously highlighted, the verdict is still out on the influence of COMT polymorphisms on L-dopa efficacy. Genetic variability of the COMT enzyme has demonstrated at least some influence on the dosing of L-dopa, which may be useful in designing individualized treatment regimens. Stronger pharmacogenetic associations have emerged as they pertain to L-dopa-induced adverse effects, but the body of evidence is still inadequate to make definitive assertions. What is obvious from these investigations is that the clinical implications of such genetic variability on L-dopa therapy remain inconclusive with respect to therapeutic response and limited with regard to drug toxicity.
Monoamine Oxidase Type B Inhibitors
MAO-B is the primary enzyme in the brain responsible for the breakdown of dopamine. MAO-B inhibitors are used concomitantly with L-dopa therapy to potentiate the effects of L-dopa. This combination therapy results in both beneficial dopaminergic effects on motor function and the potential for the dose reductions of L-dopa, which may help to decrease rates of significant adverse events. Currently, there are two FDA-approved MAO-B inhibitors indicated for the treatment of PD—selegiline and rasagiline. Selegiline is a selective, noncompetitive antagonist of the MAO-B enzyme and is indicated as an adjuvant agent to L-dopa therapy. Rasagiline is a selective, irreversible inhibitor of MAO-B with 5–10 times greater potency than selegiline and is therefore indicated for monotherapy in early stages of PD.109
Genetic polymorphisms of the cytochrome P450 superfamily of enzymes have been postulated to play a role influencing treatment of PD with MAO-B inhibitors. Selegiline has been found to be associated with gene polymorphisms of the CYP450 superfamily of enzymes, most notably CYP2B6. CYP2B6 had long been regarded a minor contributor in drug metabolism. In recent years, however, it has been discovered that CYP2B6 makes up approximately 1–10% of hepatic CYP content and is the major metabolizing enzyme for numerous drugs, including selegiline.110 Furthermore, it has been noted that there is up to a 250-fold interindividual variation in CYP2B6 expression due to genetic polymorphisms with 29 allelic variants identified to date (CYP2B6*1–*29).111,112 There have been several important allelic variants that have been identified that alter the level of activity of CYP2B6, although most do not translate into changes in enzymatic function. In an in vitro study performed by Watanabe et al., several allelic variants were found to affect the metabolism of selegiline.113 Two allelic variants—CYP2B6*10 and CYP2B6*14—appeared to have significantly lower enzymatic activity for selegiline N-demethylation compared with the wild-type allele.
It has been proposed that rasagiline may be associated with gene polymorphisms in CYP1A2, MAO-B, and the B-cell lymphoma 2 (Bcl-2).114 Rasagiline is metabolized by CYP1A2 and thereby might be thought to be affected by genetic polymorphisms in this enzyme. To date, however, no allelic variants in CYP1A2 have been found to affect treatment with rasagiline, and genetic polymorphisms in BCL-2 and MAO-B have not been studied to any extent with relation to rasagiline therapy.
The knowledge regarding the effect of genetic variability on MAO-B inhibitors is still in its infancy and it is not yet known exactly how these genetic variations will affect treatment efficacy and toxicity. The available information regarding the effect of genetic polymorphisms on the metabolism of selegiline is limited, but the findings thus far warrant future studies to investigate the clinical implications. Considering the important role CYP2B6 plays in selegiline metabolism, genetic polymorphisms of this enzyme may prove to have some impact on drug exposure and clinical effectiveness in PD patients. More research is needed to evaluate the role of genetic polymorphism on rasagiline therapy, and this will most likely focus on variations of the CYP1A2, MAOB, and BCL2 genes.
Catechol-O-Methyltransferase Inhibitors
COMT is an enzyme that functions alongside DOPA decarboxylase to convert L-dopa to a less active metabolite, 3-O-methyldopa (3-OMD). COMT is found to its greatest extent in the liver and kidneys but is also present throughout other peripheral sites and centrally in neuronal tissues. COMT inhibitors—entacapone and tolcapone—are another class of medications used as an adjunct to L-dopa therapy in the treatment of PD. They are utilized to extend the half-life and duration of action of L-dopa, which also may allow the use of smaller L-dopa dose for therapeutic effect. Entacapone is reversible COMT inhibitor that acts only in the periphery and is more often used specifically for patients who experience “wearing off” symptoms associated with long-term L-dopa treatment. Tolcapone is a selective, reversible COMT inhibitor that acts at both peripheral and central sites and, thus, it is more potent and longer-acting than entacapone. However, tolcapone has been issued a black box warning for possible hepatotoxicity while entacapone has not, making the latter agent the preferred COMT inhibitor.115,116
Genetic polymorphisms of the genes that encode COMT and UDP-glucuronosyltransferase 1A9 (UGT1A9) have been studied with respect to the COMT inhibitors. The Val158 Met polymorphism implicated in L-dopa therapy has also been thought to contribute to the efficacy of COMT inhibitors. Lee et al. investigated the effect of different COMT genotypes on the therapeutic efficacy of entacapone but concluded that COMT genotype was at best a minor factor contributing to clinical response.117 Conversely, Corvol et al. found that COMTH/H and COMTL/L polymorphisms may in fact play a role in affecting the clinical response of L-dopa–entacapone therapy.118 Their investigation was a randomized crossover clinical trial in 33 PD patients, each genotyped for COMT polymorphism, with the primary objective of assessing the gain in “on” time (i.e., period when symptoms are alleviated). The gain in “on” time in the COMTH/H group was significantly higher than that in the COMTL/L group (39 ± 10 minutes vs. 9 ± 9 minutes, ![]() ). The authors also observed a significant interaction between COMT genotype and the response to entacapone on the pharmacokinetics of L-dopa (
). The authors also observed a significant interaction between COMT genotype and the response to entacapone on the pharmacokinetics of L-dopa (![]() ).
).
Both entacapone and tolcapone are eliminated by glucuronidation through UGT1A9. Genetic mutations associated with UGT1A9 have been shown to affect glucuronidation of other drugs.119,120 Allelic variants of UGT1A9 have been less studied as it pertains to COMT inhibitors, but they may play a critical role in tolcapone-induced hepatotoxicity. There have been two case reports of female PD patients receiving tolcapone, both geno-typed to be UGT1A9 PM, who developed severe hepatocellular injury.121 These cases of hepatotoxicity prompted the FDA to issue a black box warning and require more stringent monitoring parameters for tolcapone.122
As seen with the boxed warning issued for tolcapone hepatotoxicity, genetic polymorphisms of UGT1A9 hold great potential to shape the way we approach PD treatment using COMT inhibitors. Conflicting results regarding COMT polymorphism, however, underlie the importance of more replicative or confirmatory investigations to establish a clinically relevant link to PD pharmacotherapy.
Dopamine Agonists
Dopamine agonists include bromocriptine, pramipexole, and ropinirole, all of which are approved for monotherapy or in combination with L-dopa. Use as monotherapy may extend the time before L-dopa therapy is required. Use as an adjuvant drug allows for reduction in the L-dopa dose.
Bromocriptine is an ergot derivative that acts as an agonist on dopamine D2 receptors and an antagonist on dopamine D1 receptors in the CNS. It has been shown to be a substrate for the efflux transporter P-gp at the BBB in mice, and efficacy of bromocriptine is thought to be associated with genetic polymorphisms in the ABCB1 gene, which encodes P-gp.114,123 It is suggested that changes in P-gp expression or efflux activity as a consequence of genetic polymorphisms would significantly alter bromocriptine disposition in the brain, hence affecting the efficacy and toxicity. Over 100 allelic variants have been identified thus far in the ABCB1 gene and have been found to alter the activity levels of the efflux transporter.124 However, there have been no studies to date specifically evaluating the influence of allelic variants of ABCB1 on bromocriptine efficacy and toxicity in PD.
Pramipexole is also an ergot derivative and has been found to have large interpatient variability in therapeutic response and rates of adverse reactions. This variation in efficacy is thought to be a result of SNPs in the dopamine receptor genes DRD2 and DRD3, specifically the DRD2 TaqIA and DRD3 Ser9Gly variants. A prospective study by Liu et al. of 30 Chinese PD patients administered pramipexole investigated DRD2 TaqIA gene polymorphisms (allelic variances A1 and A2) and DRD3 Ser9Gly gene polymorphisms (allelic variances Ser/Ser, Ser/Gly, and Gly/Gly) on treatment response.125 No significant associations were observed with the DRD2 genotypes, but patients homozygous for Ser/Ser were found to have a statistically significant higher response to pramipexole over patients with the Gly allele ![]() .
.
Genetic variations in dopamine receptor D3 may indeed explain variability in pramipexole treatment efficacy, and more studies are needed to determine whether this holds true. The hypothetical impact of different ABCB1genotypes on bromocriptine efficacy and toxicity seems worthwhile to pursue, considering that many ABCB1 allelic variants have been shown to affect P-gp activity. Again, additional studies are necessary to evaluate whether genetic polymorphisms will affect decisions in the treatment of PD patients with dopamine agonists.
NMDA Receptor Antagonist
Amantadine is the only NMDA receptor antagonist approved for use as monotherapy and as an adjuvant agent for the treatment of PD. Its exact mechanism has not been fully elucidated, but it is thought to potentiate dopaminergic responses that in turn increases dopamine release and inhibits reuptake.126 These actions are independent of its antiviral effects. Amantadine has been found to be associated with polymorphisms in the genes that encode dopamine receptors D1 and D2, but no studies to date have been undertaken to correlate this with efficacy or tolerability of the drug.114
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


