1
Pharmacodynamics
This chapter will be most useful after having a basic understanding of the material in Chapter 3, Pharmacodynamics: Molecular Mechanisms of Drug Action in Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 12th Edition. Additional information related to this chapter is provided in Chapter 1, Drug Invention and the Pharmaceutical Industry and Chapter 7, Pharmacogenetics. The drugs presented in this chapter are used to illustrate general pharmacodynamic principles. The mechanisms of action and therapeutic uses of drugs described in this chapter are discussed in more detail in subsequent chapters. Neither a Mechanisms of Action Table nor a Clinical Summary Table is included in this chapter because this information is provided in subsequent chapters.
In addition to the material presented here, Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 12th Edition contains:
• A description in Chapter 1 of the process of drug invention and FDA approval
• Table 1-1, Typical Characteristics of the Various Phases of the Clinical Trials Required for Marketing of New Drugs
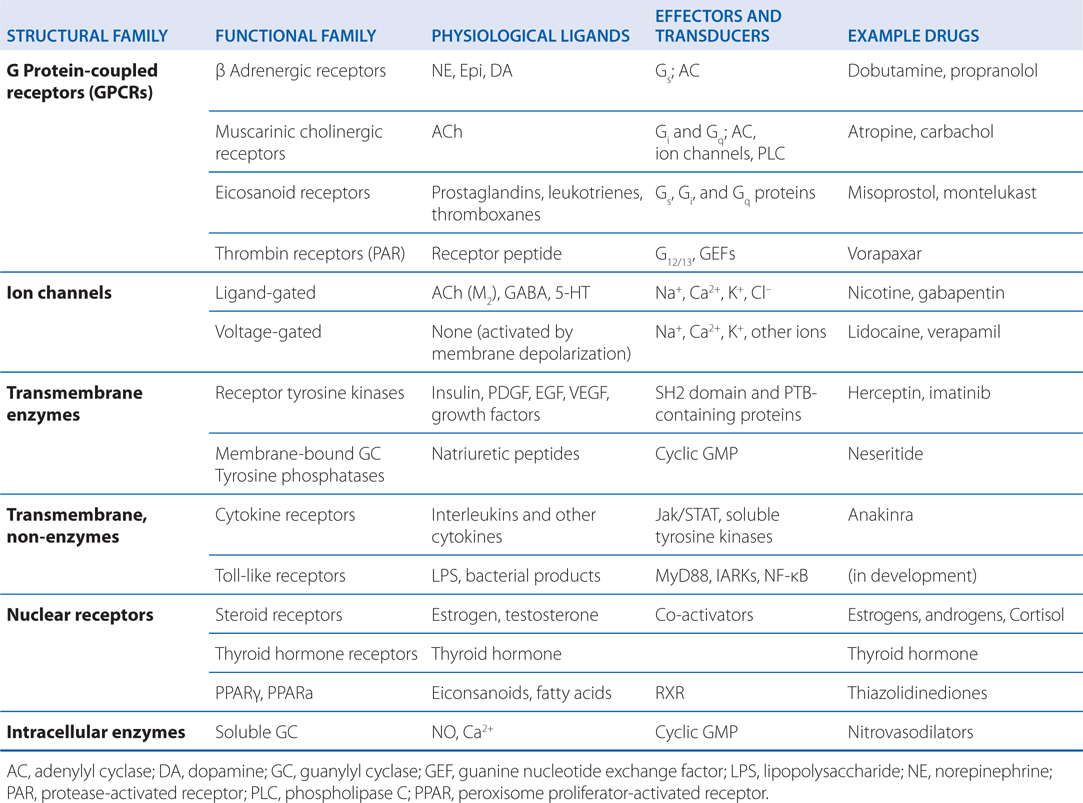
TABLE 1-1 Physiological Receptors
• Figure 1-1, The Phases, Time Lines, and Attrition That Characterize the Invention of New Drugs
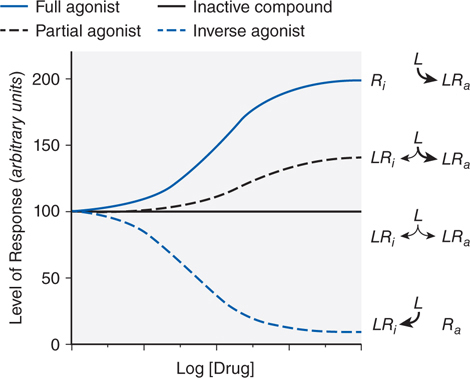
FIGURE 1-1 Regulation of the activity of a receptor with conformation-selective drugs. The ordinate is the activity of the receptor produced by Ra, the active receptor conformation (eg, stimulation of adenylyl cyclase by a β adrenergic receptor). If a drug L selectively binds to Ra, it will produce a maximal response. If L has equal affinity for Ri and Ra, it will not perturb the equilibrium between them and will have no effect on net activity; L would appear as an inactive compound. If the drug selectively binds to Ri, then the net amount of Ra will be diminished. If L can bind to receptor in an active conformation Ra but also bind to inactive receptor Ri with lower affinity, the drug will produce a partial response; L will be a partial agonist. If there is sufficient Ra to produce an elevated basal response in the absence of ligand (agonist-independent constitutive activity), then activity will be inhibited; L will then be an inverse agonist. Inverse agonists selectively bind to the inactive form of the receptor and shift the conformational equilibrium toward the inactive state. In systems that are not constitutively active, inverse agonists will behave like competitive antagonists, which helps explain why the properties of inverse agonists and the number of such agents previously described as competitive antagonists were only recently appreciated. Receptors that have constitutive activity and are sensitive to inverse agonists include benzodiazepine, histamine, opioid, cannabinoid, dopamine, bradykinin, and adenosine receptors.
• A comprehensive description in Chapter 3 of the various mechanisms of drug action, including cellular pathways activated by physiological receptors, structural and functional families of physiological receptors, second messengers, ion channels, nuclear receptors, and transcription factors
• Chapter 3 also describes mechanisms of receptor desensitization and regulation, and an example of pharmacodynamic interactions in a multicellular context
• Chapter 7 includes a number of examples of genetic polymorphisms that affect drug pharmacodynamics, the impact of pharmacogenetics on drug development, and a discussion of pharmacogenetics in clinical practice
LEARNING OBJECTIVES
 Understand key concepts and terms related to pharmacodynamics, including drug receptor agonism and antagonism.
Understand key concepts and terms related to pharmacodynamics, including drug receptor agonism and antagonism.
 Know concepts and terms that are used to quantify drug receptor interactions, including affinity, efficacy, potency, KD and Ki.
Know concepts and terms that are used to quantify drug receptor interactions, including affinity, efficacy, potency, KD and Ki.
 Understand how drug pharmacodynamic information is used to predict beneficial and toxic drug effects.
Understand how drug pharmacodynamic information is used to predict beneficial and toxic drug effects.
 Know the modern process of drug invention and FDA approval, and post-market surveillance.
Know the modern process of drug invention and FDA approval, and post-market surveillance.
 Know how genetic polymorphisms and other factors can affect the pharmacodynamic properties of drugs and lead to variability in individual patient responses to drugs.
Know how genetic polymorphisms and other factors can affect the pharmacodynamic properties of drugs and lead to variability in individual patient responses to drugs.
 Know how pharmacodynamics is applied to populations of patients to estimate population therapeutic windows for drug dosing.
Know how pharmacodynamics is applied to populations of patients to estimate population therapeutic windows for drug dosing.
PHARMACODYNAMIC CONCEPTS AND NOMENCLATURE (key terms are indicated in italics)
• Pharmacodynamics is the study of the biochemical and physiological effects of drugs and their mechanisms of action.
• Pharmacodynamics refers to the effects of a drug on the body; in contrast, the effects of the body on the actions of a drug are pharmacokinetic processes (see Chapter 2).
• The term drug receptor or drug target denotes the cellular macromolecule or macromolecular complex with which the drug interacts to elicit a cellular response.
• Drugs commonly alter the rate or magnitude of an intrinsic cellular response rather than create new responses.
• Drug receptors are often located on the surface of cells but may also be located in specific intracellular compartments.
• Many drugs also interact with acceptors within the body; acceptors are entities that do not directly cause any change in biochemical or physiological response.
• Proteins form the most important class of drug receptors. Examples include:
▶ Physiological receptors for hormones, growth factors, transcription factors, and neurotransmitters (see Table 1-1)
▶ Enzymes of crucial metabolic or regulatory pathways (eg, dihydrofolate reductase, acetylcholinesterase, and cyclic nucleotide phosphodiesterases)
▶ Proteins involved in transport processes (eg, Na+, K+-ATPase); secreted glycoproteins (eg, Wnts)
▶ Structural proteins (eg, tubulin)
• Specific binding of drugs to other cellular constituents such as DNA is also exploited for therapeutic purposes (eg, many cancer chemotherapeutic agents and antiviral drugs).
• Antibiotic and other anti-infectives often target enzymes and biochemical processes that are unique to the pathogen, resulting in cytotoxicity or inhibition of proliferation.
A man who suffers from hay fever is at his local pharmacy looking for a nonprescription drug that can provide relief of symptoms. One of the allergy medicines he finds on the shelf says the active ingredient is an antihistamine, diphenhydramine.
a. What is an “active ingredient” in an allergy medication such as this?
Pharmaceuticals contain 1 or more pharmacologically active ingredients, which are termed active ingredients. The quantities of each active ingredient in each dose (eg, tablet, capsule, or volume of a liquid medication) are indicated on the packaging. In addition to active ingredients, most medications also contain other ingredients that are pharmacologically inactive, but may improve the pharmacokinetics, appearance, taste, shelf-life, or other properties that may enhance dosing and product effectiveness.
b. What kind of drug is an “antihistamine” and why is it so named?
As the name indicates, an antihistamine is an agent that antagonizes the action of the endogenous mediator histamine. Histamine is released by mast cells and is involved in a variety of allergic and inflammatory responses (see Chapter 21). This man’s hay fever is caused by mast cell release of histamine and other substances that result in his symptoms of sneezing, runny nose, itching of the nose and throat, and itchy, watery eyes. An antihistamine such as diphenhydramine will bind to histamine receptors on cells throughout the body and block the effects of histamine.
DRUG-RECEPTOR INTERACTIONS (key terms are indicated in italics)
• Many drugs act on physiological receptors and are particularly selective because physiological receptors have evolved to recognize and respond to individual signaling molecules with great selectivity.
• Drugs that bind to physiological receptors and mimic the regulatory effects of the endogenous signaling compounds are termed agonists.
▶ A drug that binds to the same recognition site as the endogenous agonist (the primary or orthosteric site on the receptor) is said to be a primary agonist.
▶ Drugs that can elicit the same maximal response as the endogenous ligand are termed full agonists (see Figure 1-1) and are said to possess full efficacy.
▶ Drugs that are only partly effective in activating a receptor regardless of the concentration employed are termed partial agonists (see Figure 1-1).
▶ A syntopic interaction is an interaction between ligands that bind to the same recognition site, or to recognition sites that overlap, on the receptor macromolecule (see Figure 1-2).
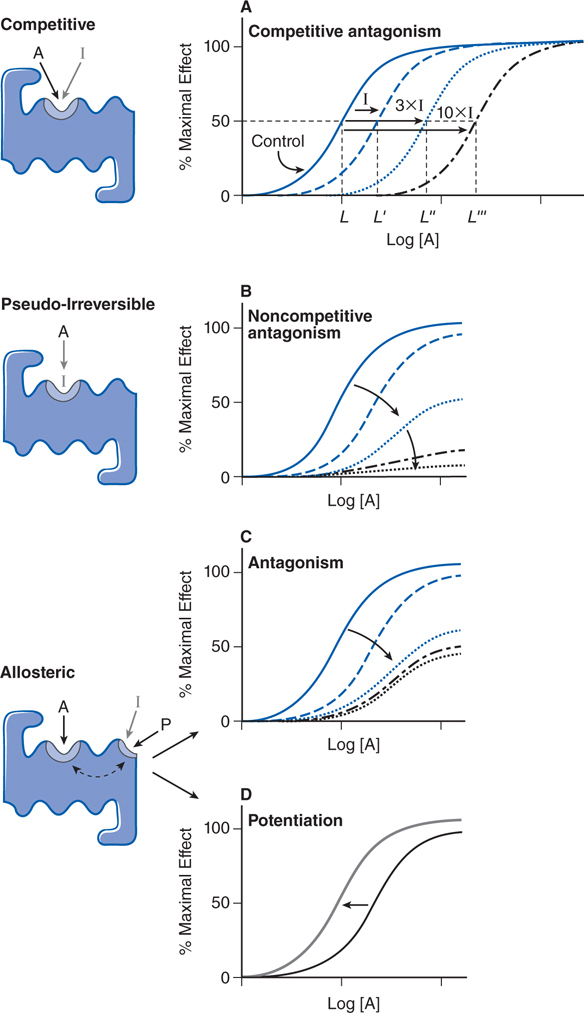
FIGURE 1-2 Mechanisms of receptor antagonism and potentiation. A. Competitive antagonism occurs when the agonist A and antagonist I compete for the same binding site on the receptor. Response curves for the agonist are shifted to the right in a concentration-related manner by the antagonist such that the EC50 for the agonist increases (eg, L versus L′, L″, and L′″) with the concentration of the antagonist. B. If the antagonist binds to the same site as the agonist but does so irreversibly or pseudo-irreversibly (slow dissociation but no covalent bond), it causes a shift of the dose-response curve to the right, with further depression of the maximal response. Allosteric effects occur when an allosteric ligand I or P binds to a different site on the receptor to either inhibit (I) the response (see panel C) or potentiate (P) the response (see panel D). This effect is saturable; inhibition or potentiation reaches a limiting value when the allosteric site is fully occupied.
▶ Allosteric (allotopic) agonists bind to an allosteric or allotopic site, a region on the receptor that is different from the primary site (see Figure 1-2).
• Many receptors exhibit some constitutive activity in the absence of a regulatory ligand; drugs that stabilize such receptors in an inactive conformation are termed inverse agonists (see Figure 1-1).
• Drugs that block or reduce the action of an agonist are termed antagonists (see Figure 1-2).
▶ Antagonism most commonly results from competition of a drug with an agonist for the primary site on the receptor (referred to as competitive antagonists because of a syntopic interaction; see Figure 1-2).
▶ Noncompetitive antagonism results from an antagonist that covalently binds the receptor or dissociates extremely slowly from the receptor (pseudo-irreversible) such that the maximal response of the receptor is diminished with increasing concentration of antagonist (see Figure 1-2).
▶ Noncompetitive antagonism can also occur with antagonists that interact with allosteric sites on the receptor (allosteric antagonists), reducing the affinity of the receptor for agonist.
▶ Drugs that act by combining with the agonist are termed chemical antagonists.
▶ Functional antagonists act by indirectly inhibiting the cellular or physiological effects of the agonist.
• Partial agonists and inverse agonists that compete with a full agonist at the same site on a receptor (ie, syntopic interaction) will behave as competitive antagonists.
• Specificity is a pharmacodynamic property of a drug that refers to a drug’s ability to elicit a well-defined response in a specific tissue.
▶ The chemical structure of a drug contributes to its specificity for a given type of physiological receptor.
▶ A drug that interacts with a single type of receptor that is expressed on only a limited number of differentiated cells will exhibit high (narrow) specificity.
▶ Drugs acting on a type of receptor that is expressed ubiquitously on a variety of cells throughout the body will exhibit widespread effects, and could produce serious side effects or toxicities if the receptor serves important functions in multiple tissues.
▶ Many clinically important drugs exhibit a low (broad) specificity because the drug is able to interact with multiple receptors in different tissues; such broad specificity might enhance the clinical utility of a drug, but also contribute to a spectrum of adverse side effects due to off-target interactions.
c. What are histamine receptors and what kind of an antagonist is diphenhydramine?
Histamine receptors are G protein–coupled receptors (GPCRs; see Table 1-1). H1 receptors couple to Gq/11 and activate the PLC–IP3–Ca2+ pathway and its many possible sequelae, including activation of PKC, Ca2+–calmodulin–dependent enzymes (eNOS and various protein kinases), and PLA2. H2 receptors link to Gs to activate the adenylyl cyclase–cyclic AMP–PKA pathway, whereas H3 and H4 receptors couple to Gi/o to inhibit adenylyl cyclase and decrease cellular cyclic AMP. Activation of H3 receptors also can activate mitogen-activated protein (MAP) kinase and inhibit the Na+/H+ exchanger, and activation of H4 receptors mobilizes stored Ca2+ in some cells.
All the available H1 receptor “antagonists” are actually inverse agonists (see Side Bar DRUG-RECEPTOR INTERACTIONS and Figure 1-1) that reduce constitutive activity of the receptor and compete with histamine. Whereas histamine binding to the receptor induces a fully active conformation, antihistamine binding yields an inactive conformation (see Figure 1-1). At the tissue level, the effect seen is proportional to receptor occupancy by the antihistamine.
d. The warning on the medication package says “marked drowsiness may occur.” The package label also warns that “excitability may occur, especially in children.” How are these side effects related to the “antihistamine” effects of the active ingredient?
Some antihistamines such as diphenhydramine can cross the blood-brain barrier (BBB) and have effects on histamine receptors in the central nervous system (CNS). There is substantial evidence that histamine functions as a neurotransmitter in the CNS. Histamine, histidine decarboxylase, enzymes that metabolize histamine, and H1, H2, and H3 receptors are distributed widely but nonuniformly in the CNS. Histamine-containing neurons control both homeostatic and higher brain functions, including regulation of the sleep-wake cycle, circadian and feeding rhythms, immunity, learning, memory, drinking, and body temperature.
The first-generation H1 antagonists can both stimulate and depress the CNS. Stimulation occasionally is encountered in patients given conventional doses; they become restless, nervous, and unable to sleep. Central excitation also is a striking feature of overdose, which commonly results in convulsions, particularly in infants. Central depression, on the other hand, usually accompanies therapeutic doses of the older H1 antagonists. Diminished alertness, slowed reaction times, and somnolence are common manifestations. Patients vary in their susceptibility and responses to individual drugs. The ethanolamines (eg, diphenhydramine) are particularly prone to causing sedation. Thus, the development of second-generation “nonsedating” antihistamines was an important advance that allowed their general use. These newer H1 antagonists do not cross the blood-brain barrier appreciably. Their sedative effects are similar to those of placebo.
e. Are there other pharmacological properties of H1 antagonists that are clinically useful?
Many of the first-generation H1 antagonists tend to inhibit responses to acetylcholine (ACh) that are mediated by muscarinic receptors and may be manifest during clinical use (see Chapter 6). Some H1 antagonists also can be used to treat motion sickness; the anticholinergic properties of H1 antagonists may be largely responsible for this effect. Indeed, promethazine (a first-generation H1 antagonist) has perhaps the strongest muscarinic-blocking activity among these agents and is the most effective H1 antagonist in combating motion sickness. The second-generation H1 antagonists have no effect on muscarinic receptors.
A pharmaceutical company research and development team is working to develop new drugs that target a novel molecular entity that might play a role in diabetes. Preclinical data regarding the pharmacodynamics of a number of small molecules have been collected and are being analyzed to determine which lead compounds might be worth carrying forward into clinical trials in humans.
a. What are the steps that the team has taken to this point and what must be done before the drug is tested in humans?
Modern drug invention usually starts with a statement (or hypothesis) that a certain protein or pathway plays a critical role in the pathogenesis of a certain disease, and that altering the protein’s activity would therefore be effective against that disease. The usual approach to invention of a small-molecule drug is to screen a collection of chemicals (“library”) for compounds with the desired features.
QUANTITATIVE ASPECTS OF DRUG RECEPTOR INTERACTIONS
• Receptor occupancy theory assumes that response emanates from a receptor occupied by a drug, a concept that has its basis in the law of mass action.
• The dose-response (or concentration-response) curve is a depiction of the observed effect of a drug as a function of its concentration in the receptor compartment (see Figure 1-3).
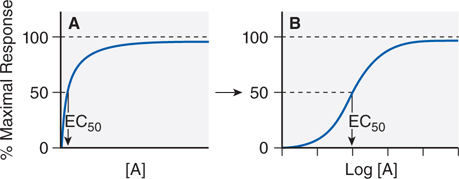
FIGURE 1-3 Graded responses (y axis as a percentage of maximal response) expressed as a function of the concentration of drug A present at the receptor. The hyperbolic shape of the curve in panel A becomes sigmoid when plotted semi-logarithmically, as in panel B. The concentration of drug that produces 50% of the maximal response quantifies drug activity and is referred to as the EC50 (effective concentration for 50% response). The range of concentrations needed to fully depict the dose-response relationship (~3 log10 units) is too wide to be useful in the linear format of Figure 1-3A; thus, most dose-response curves use log [Drug] on the x axis, as in Figure 1-3B. Dose-response curves presented in this way are sigmoidal in shape and have 3 properties: threshold, slope, and maximal asymptote. These 3 parameters quantitate the activity of the drug.
▶ The maximal asymptotic response occurs when the drug occupies all the receptor sites.
▶ Some drug-receptor systems exhibit an inverted U-shaped dose-response relationship with low-dose stimulation and high-dose inhibition of response, an effect known as hormesis.
• The bimolecular drug-receptor interaction that results in receptor activation can be described as a series of 2 equilibrium reactions as shown in Equation 1-1.
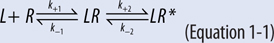
• The initial reaction is the reversible binding of drug (L) to a receptor (R) which leads to the formation of the drug-receptor complex (LR), and depends on both the forward or association rate (k+1) and the reverse or dissociation rate (k–1).
▶ The equilibrium dissociation constant (KD) is defined as the ratio of the off and on rate constants (k–1/k+1; Equation 1-2); the affinity or equilibrium association constant (KA) is the reciprocal of the equilibrium dissociation constant.
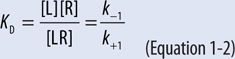
▶ Thus a high-affinity drug has a low KD and will bind a greater number of a particular receptor at a low concentration than a low-affinity drug.
▶ Depending on the characteristics of the drug (eg, full agonist, partial agonist, antagonist, inverse agonist; see Figure 1-1), the drug-receptor complex may undergo a conformational change that leads to the formation of the activated complex (LR*).
▶ The apparent dissociation constant, Kapp, is a macroscopic equilibrium constant that reflects both the ligand binding equilibrium and the subsequent equilibrium that results in the formation of the active receptor LR*.
▶ The fractional occupancy of a receptor, f, is described by Equations 1-3 and 1-4; at a concentration of drug that results in 50% occupancy of the receptor (f = 0.5), the concentration of drug equals the KD.

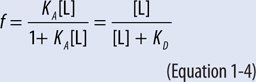
▶ The concentration of an agonist drug that produces 50% of the maximal response is termed the EC50 (the half-maximally effective concentration; see Figure 1-3).
• The ability of a drug to activate a receptor and generate a cellular response is a reflection of its efficacy; relative efficacy refers to a comparison of the maximal response of 2 agonists; the more efficacious drug exhibits a higher maximal response (see Figure 1-4).
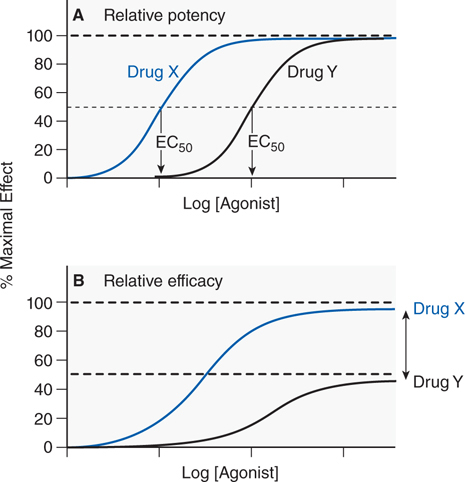
FIGURE 1-4 Two ways of quantifying agonism. A. The relative potency of 2 agonists (Drug X, blue line; Drug Y, black line) obtained in the same tissue is a function of their relative affinities and intrinsic efficacies. The EC50 of Drug X occurs at a concentration that is one-tenth the EC50 of Drug Y. Thus, Drug X is more potent than Drug Y. B. In systems where the 2 drugs do not both produce the same maximal response characteristic of the tissue, the observed maximal response is a nonlinear function of their relative intrinsic efficacies. Drug X is more efficacious than Drug Y; their asymptotic fractional responses are 100% (Drug X) and 50% (Drug Y).
▶ A full agonist has full efficacy; a partial agonist has partial efficacy.
• Drug potency refers to a comparison of the concentrations of agonist drug required to elicit a given biological response and is a mixed function of both affinity and efficacy.
▶ In comparing the EC50 of 2 drugs with the same efficacy, the drug with the lower EC50 is the more potent drug (see Figure 1-4).
• The affinity of a competitive antagonist (Ki) for its receptor can be determined in radioligand binding assays or by measuring the functional response of a system to a drug in the presence of the antagonist (see text and Equations 3-3 and 3-4 in Chapter 3 of Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 12th Edition).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


