395 | Gout and Other Crystal-Associated Arthropathies |
The use of polarizing light microscopy during synovial fluid analysis in 1961 by McCarty and Hollander and the subsequent application of other crystallographic techniques, such as electron microscopy, energy-dispersive elemental analysis, and x-ray diffraction, have allowed investigators to identify the roles of different microcrystals, including monosodium urate (MSU), calcium pyrophosphate (CPP), calcium apatite (apatite), and calcium oxalate (CaOx), in inducing acute or chronic arthritis or periarthritis. The clinical events that result from deposition of MSU, CPP, apatite, and CaOx have many similarities but also have important differences. Because of often similar clinical presentations, the need to perform synovial fluid analysis to distinguish the type of crystal involved must be emphasized. Polarized light microscopy alone can identify most typical crystals; apatite, however, is an exception. Aspiration and analysis of effusions are also important to assess the possibility of infection. Apart from the identification of specific microcrystalline materials or organisms, synovial fluid characteristics in crystal-associated diseases are nonspecific, and synovial fluid can be inflammatory or noninflammatory. Without crystal identification, these diseases can be confused with rheumatoid or other types of arthritis. A list of possible musculoskeletal manifestations of crystal-associated arthritis is shown in Table 395-1.
MUSCULOSKELETAL MANIFESTATIONS OF CRYSTAL-INDUCED ARTHRITIS |
GOUT
Gout is a metabolic disease that most often affects middle-aged to elderly men and postmenopausal women. It results from an increased body pool of urate with hyperuricemia. It typically is characterized by episodic acute arthritis or chronic arthritis caused by deposition of MSU crystals in joints and connective tissue tophi and the risk for deposition in kidney interstitium or uric acid nephrolithiasis (Chap. 431e).
ACUTE AND CHRONIC ARTHRITIS
Acute arthritis is the most common early clinical manifestation of gout. Usually, only one joint is affected initially, but polyarticular acute gout can occur in subsequent episodes. The metatarsophalangeal joint of the first toe often is involved, but tarsal joints, ankles, and knees also are affected commonly. Especially in elderly patients or in advanced disease, finger joints may be involved. Inflamed Heberden’s or Bouchard’s nodes may be a first manifestation of gouty arthritis. The first episode of acute gouty arthritis frequently begins at night with dramatic joint pain and swelling. Joints rapidly become warm, red, and tender, with a clinical appearance that often mimics that of cellulitis. Early attacks tend to subside spontaneously within 3–10 days, and most patients have intervals of varying length with no residual symptoms until the next episode. Several events may precipitate acute gouty arthritis: dietary excess, trauma, surgery, excessive ethanol ingestion, hypouricemic therapy, and serious medical illnesses such as myocardial infarction and stroke.
After many acute mono- or oligoarticular attacks, a proportion of gouty patients may present with a chronic nonsymmetric synovitis, causing potential confusion with rheumatoid arthritis (Chap. 380). Less commonly, chronic gouty arthritis will be the only manifestation, and, more rarely, the disease will manifest only as periarticular tophaceous deposits in the absence of synovitis. Women represent only 5–20% of all patients with gout. Most women with gouty arthritis are postmenopausal and elderly, have osteoarthritis and arterial hypertension that causes mild renal insufficiency, and usually are receiving diuretics. Premenopausal gout is rare. Kindreds of precocious gout in young females caused by decreased renal urate clearance and renal insufficiency have been described.
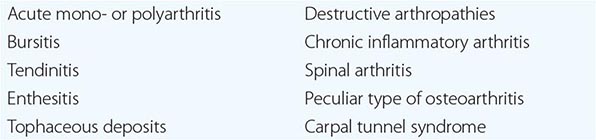
Laboratory Diagnosis Even if the clinical appearance strongly suggests gout, the presumptive diagnosis ideally should be confirmed by needle aspiration of acutely or chronically involved joints or tophaceous deposits. Acute septic arthritis, several of the other crystalline-associated arthropathies, palindromic rheumatism, and psoriatic arthritis may present with similar clinical features. During acute gouty attacks, needle-shaped MSU crystals typically are seen both intracellularly and extracellularly (Fig. 395-1). With compensated polarized light, these crystals are brightly birefringent with negative elongation. Synovial fluid leukocyte counts are elevated from 2000 to 60,000/μL. Effusions appear cloudy due to the increased numbers of leukocytes. Large amounts of crystals occasionally produce a thick pasty or chalky joint fluid. Bacterial infection can coexist with urate crystals in synovial fluid; if there is any suspicion of septic arthritis, joint fluid must be cultured.
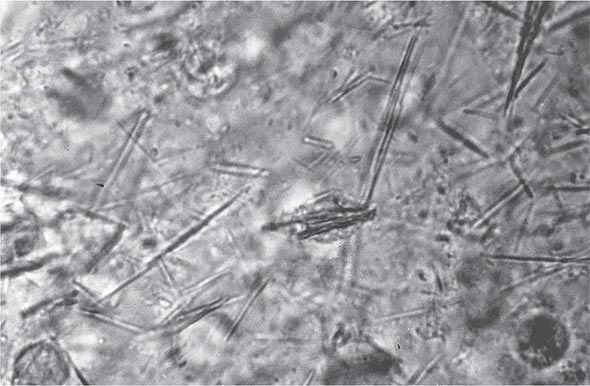
FIGURE 395-1 Extracellular and intracellular monosodium urate crystals, as seen in a fresh preparation of synovial fluid, illustrate needle-and rod-shaped crystals. These crystals are strongly negative birefringent crystals under compensated polarized light microscopy; 400×.
MSU crystals also can often be demonstrated in the first metatarsophalangeal joint and in knees not acutely involved with gout. Arthrocentesis of these joints is a useful technique to establish the diagnosis of gout between attacks.
Serum uric acid levels can be normal or low at the time of an acute attack, as inflammatory cytokines can be uricosuric and effective initiation of hypouricemic therapy can precipitate attacks. This limits the value of serum uric acid determinations for the diagnosis of gout. Nevertheless, serum urate levels are almost always elevated at some time and are important to use to follow the course of hypouricemic therapy. A 24-h urine collection for uric acid can, in some cases, be useful in assessing the risk of stones, elucidating overproduction or underexcretion of uric acid, and deciding whether it may be appropriate to use a uricosuric therapy (Chap. 431e). Excretion of >800 mg of uric acid per 24 h on a regular diet suggests that causes of overproduction of purine should be considered. Urinalysis, serum creatinine, hemoglobin, white blood cell (WBC) count, liver function tests, and serum lipids should be obtained because of possible pathologic sequelae of gout and other associated diseases requiring treatment and as baselines because of possible adverse effects of gout treatment.
Radiographic Features Cystic changes, well-defined erosions with sclerotic margins (often with overhanging bony edges), and soft tissue masses are characteristic radiographic features of advanced chronic tophaceous gout. Ultrasound may aid earlier diagnosis by showing a double contour sign overlying the articular cartilage. Dual-energy computed tomography (CT) can show specific features establishing the presence of urate crystals.
CALCIUM PYROPHOSPHATE DEPOSITION (CPPD) DISEASE
PATHOGENESIS
The deposition of CPP crystals in articular tissues is most common in the elderly, occurring in 10–15% of persons age 65–75 years and 30–50% of those >85 years. In most cases, this process is asymptomatic and the cause of CPPD is uncertain. Because >80% of patients are >60 years and 70% have preexisting joint damage from other conditions, it is likely that biochemical changes in aging or diseased cartilage favor crystal nucleation. In patients with CPPD arthritis, there is increased production of inorganic pyrophosphate and decreased levels of pyrophosphatases in cartilage extracts. Mutations in the ANKH gene, as described in both familial and sporadic cases, can increase elaboration and extracellular transport of pyrophosphate. The increase in pyrophosphate production appears to be related to enhanced activity of ATP pyrophosphohydrolase and 5′-nucleotidase, which catalyze the reaction of ATP to adenosine and pyrophosphate. This pyrophosphate could combine with calcium to form CPP crystals in matrix vesicles or on collagen fibers. There are decreased levels of cartilage glycosaminoglycans that normally inhibit and regulate crystal nucleation. High activities of transglutaminase enzymes also may contribute to the deposition of CPP crystals.
Release of CPP crystals into the joint space is followed by the phagocytosis of those crystals by monocyte-macrophages and neutrophils, which respond by releasing chemotactic and inflammatory substances and, as with MSU crystals, activating the inflammasome.
A minority of patients with CPPD arthropathy have metabolic abnormalities or hereditary CPP disease (Table 395-2). These associations suggest that a variety of different metabolic products may enhance CPP crystal deposition either by directly altering cartilage or by inhibiting inorganic pyrophosphatases. Included among these conditions are hyperparathyroidism, hemochromatosis, hypophosphatasia, hypomagnesemia, and possibly myxedema. The presence of CPPD arthritis in individuals <50 years old should lead to consideration of these metabolic disorders (Table 395-2) and inherited forms of disease, including those identified in a variety of ethnic groups. Genomic DNA studies performed on different kindreds have shown a possible location of genetic defects on chromosome 8q or on chromosome 5p in a region that expresses the gene of the membrane pyrophosphate channel (ANKH gene). As noted above, mutations described in the ANKH gene in kindreds with CPPD arthritis can increase extracellular pyrophosphate and induce CPP crystal formation. Investigation of younger patients with CPPD should include inquiry for evidence of familial aggregation and evaluation of serum calcium, phosphorus, alkaline phosphatase, magnesium, iron, and transferrin.
CONDITIONS ASSOCIATED WITH CALCIUM PYROPHOSPHATE CRYSTAL DEPOSITION DISEASE |
CLINICAL MANIFESTATIONS
CPPD arthropathy may be asymptomatic, acute, subacute, or chronic or may cause acute synovitis superimposed on chronically involved joints. Acute CPPD arthritis originally was termed pseudogout by McCarty and co-workers because of its striking similarity to gout. Other clinical manifestations of CPPD include (1) association with or enhancement of peculiar forms of osteoarthritis; (2) induction of severe destructive disease that may radiographically mimic neuropathic arthritis; (3) production of chronic symmetric synovitis that is clinically similar to rheumatoid arthritis; (4) intervertebral disk and ligament calcification with restriction of spine mobility, the crowned dens syndrome, or spinal stenosis (most commonly seen in the elderly); and (5) rarely periarticular tophus-like nodules.
The knee is the joint most frequently affected in CPPD arthropathy. Other sites include the wrist, shoulder, ankle, elbow, and hands. The temporomandibular joint may be involved. Clinical and radiographic evidence indicates that CPPD deposition is polyarticular in at least two-thirds of patients. When the clinical picture resembles that of slowly progressive osteoarthritis, diagnosis may be difficult. Joint distribution may provide important clues suggesting CPPD disease. For example, primary osteoarthritis less often involves metacarpophalangeal, wrist, elbow, shoulder, or ankle joints. If radiographs or ultrasound reveal punctate and/or linear radiodense deposits within fibrocartilaginous joint menisci or articular hyaline cartilage (chondrocalcinosis), the diagnostic likelihood of CPPD disease is further increased. Definitive diagnosis requires demonstration of typical rhomboid or rodlike crystals (generally weakly positively birefringent or nonbirefringent with polarized light) in synovial fluid or articular tissue (Fig. 395-2). In the absence of joint effusion or indications to obtain a synovial biopsy, chondrocalcinosis is presumptive of CPPD. One exception is chondrocalcinosis due to CaOx in some patients with chronic renal failure.
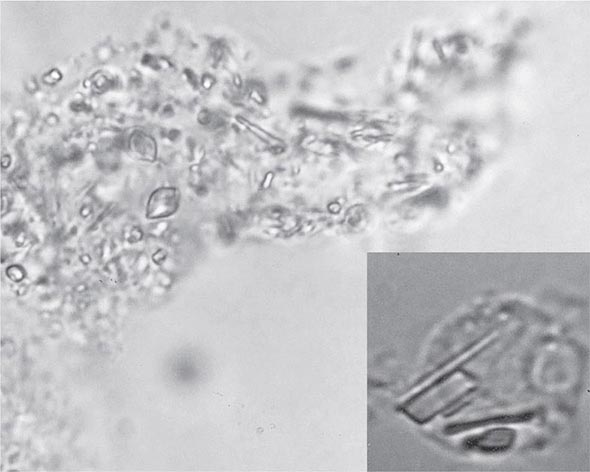
FIGURE 395-2 Intracellular and extracellular calcium pyrophosphate (CPP) crystals, as seen in a fresh preparation of synovial fluid, illustrate rectangular, rod-shaped, and rhomboid crystals that are weakly positively or nonbirefringent crystals (compensated polarized light microscopy; 400×).
Acute attacks of CPPD arthritis may be precipitated by trauma. Rapid diminution of serum calcium concentration, as may occur in severe medical illness or after surgery (especially parathyroidectomy), can also lead to attacks.
In as many as 50% of cases, episodes of CPPD-induced inflammation are associated with low-grade fever and, on occasion, temperatures as high as 40°C (104°F). In such cases, synovial fluid analysis with microbial cultures is essential to rule out the possibility of infection. In fact, infection in a joint with any microcrystalline deposition process can lead to crystal shedding and subsequent synovitis from both crystals and microorganisms. The leukocyte count in synovial fluid in acute CPPD can range from several thousand cells to 100,000 cells/μL, with the mean being about 24,000 cells/μL and the predominant cell being the neutrophil. CPP crystals may be seen inside tissue fragments and fibrin clots and in neutrophils (Fig. 395-2). CPP crystals may coexist with MSU and apatite in some cases.
CALCIUM APATITE DEPOSITION DISEASE
PATHOGENESIS
Apatite is the primary mineral of normal bone and teeth. Abnormal accumulation of basic calcium phosphates, largely carbonate substituted apatite, can occur in areas of tissue damage (dystrophic calcification), hypercalcemic or hyperparathyroid states (metastatic calcification), and certain conditions of unknown cause (Table 395-3). In chronic renal failure, hyperphosphatemia can contribute to extensive apatite deposition both in and around joints. Familial aggregation is rarely seen; no association with ANKH mutations has been described thus far. Apatite crystals are deposited primarily on matrix vessels. Incompletely understood alterations in matrix proteoglycans, phosphatases, hormones, and cytokines probably can influence crystal formation.
CONDITIONS ASSOCIATED WITH APATITE DEPOSITION DISEASE |
Abbreviation: SLE, systemic lupus erythematosus.
Apatite aggregates are commonly present in synovial fluid in an extremely destructive chronic arthropathy of the elderly that occurs most often in the shoulders (Milwaukee shoulder) and in a similar process in hips, knees, and erosive osteoarthritis of fingers. Joint destruction is associated with damage to cartilage and supporting structures, leading to instability and deformity. Progression tends to be indolent. Symptoms range from minimal to severe pain and disability that may lead to joint replacement surgery. Whether severely affected patients represent an extreme synovial tissue response to the apatite crystals that are so common in osteoarthritis is uncertain. Synovial lining cell or fibroblast cultures exposed to apatite (or CPP) crystals can undergo mitosis and markedly increase the release of prostaglandin E2, various cytokines, and also collagenases and neutral proteases, underscoring the destructive potential of abnormally stimulated synovial lining cells.
CLINICAL MANIFESTATIONS
Periarticular or articular deposits may occur and may be associated with acute reversible inflammation and/or chronic damage to the joint capsule, tendons, bursa, or articular surfaces. The most common sites of apatite deposition include bursae and tendons in and/or around the knees, shoulders, hips, and fingers. Clinical manifestations include asymptomatic radiographic abnormalities, acute synovitis, bursitis, tendinitis, and chronic destructive arthropathy. Although the true incidence of apatite arthritis is not known, 30–50% of patients with osteoarthritis have apatite microcrystals in their synovial fluid. Such crystals frequently can be identified in clinically stable osteoarthritic joints, but they are more likely to come to attention in persons experiencing acute or subacute worsening of joint pain and swelling. The synovial fluid leukocyte count in apatite arthritis is usually low (<2000/μL) despite dramatic symptoms, with predominance of mononuclear cells.
DIAGNOSIS
Intra- and/or periarticular calcifications with or without erosive, destructive, or hypertrophic changes may be seen on radiographs (Fig. 395-3). They should be distinguished from the linear calcifications typical of CPPD.
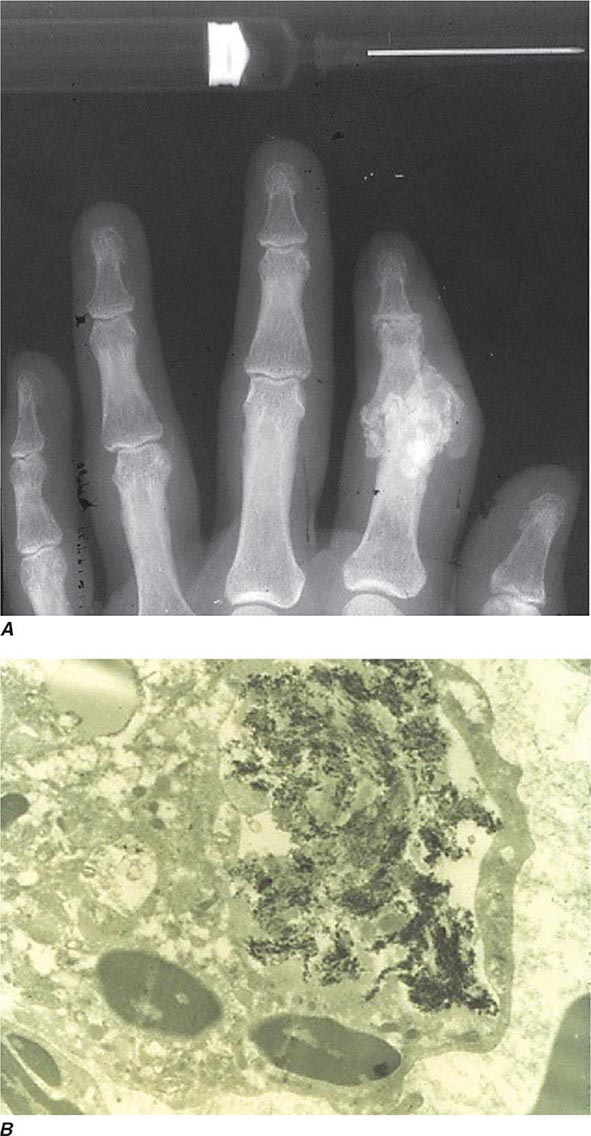
FIGURE 395-3 A. Radiograph showing calcification due to apatite crystals surrounding an eroded joint. B. An electron micrograph demonstrates dark needle-shaped apatite crystals within a vacuole of a synovial fluid mononuclear cell (30,000×).
Definitive diagnosis of apatite arthropathy, also called basic calcium phosphate disease, depends on identification of crystals from synovial fluid or tissue (Fig. 395-3). Individual crystals are very small and can be seen only by electron microscopy. Clumps of crystals may appear as 1- to 20-μm shiny intra- or extracellular nonbirefringent globules or aggregates that stain purplish with Wright’s stain and bright red with alizarin red S. Tetracycline binding and other investigative techniques are under consideration as labeling alternatives. Absolute identification depends on electron microscopy with energy-dispersive elemental analysis, x-ray diffraction, infrared spectroscopy, or Raman microspectroscopy, but these techniques usually are not required in clinical diagnosis.
CAOX DEPOSITION DISEASE
PATHOGENESIS
Primary oxalosis is a rare hereditary metabolic disorder (Chap. 434e). Enhanced production of oxalic acid may result from at least two different enzyme defects, leading to hyperoxalemia and deposition of CaOx crystals in tissues. Nephrocalcinosis and renal failure are typical results. Acute and/or chronic CaOx arthritis, periarthritis, and bone disease may complicate primary oxalosis during later years of illness.
Secondary oxalosis is more common than the primary disorder. In chronic renal disease, calcium oxalate deposits have long been recognized in visceral organs, blood vessels, bones, and cartilage and are now known to be one of the causes of arthritis in chronic renal failure. Thus far, reported patients have been dependent on long-term hemodialysis or peritoneal dialysis (Chap. 336), and many had received ascorbic acid supplements. Ascorbic acid is metabolized to oxalate, which is inadequately cleared in uremia and by dialysis. Such supplements and foods high in oxalate content usually are avoided in dialysis programs because of the risk of enhancing hyperoxalosis and its sequelae.
CLINICAL MANIFESTATIONS AND DIAGNOSIS
CaOx aggregates can be found in bone, articular cartilage, synovium, and periarticular tissues. From these sites, crystals may be shed, causing acute synovitis. Persistent aggregates of CaOx can, like apatite and CPP, stimulate synovial cell proliferation and enzyme release, resulting in progressive articular destruction. Deposits have been documented in fingers, wrists, elbows, knees, ankles, and feet.
Clinical features of acute CaOx arthritis may not be distinguishable from those due to urate, CPP, or apatite. Radiographs may reveal chondrocalcinosis or soft tissue calcifications. CaOx-induced synovial effusions are usually noninflammatory, with <2000 leukocytes/μL, or mildly inflammatory. Neutrophils or mononuclear cells can predominate. CaOx crystals have a variable shape and variable birefringence to polarized light. The most easily recognized forms are bipyramidal, have strong birefringence (Fig. 395-4), and stain with alizarin red S.
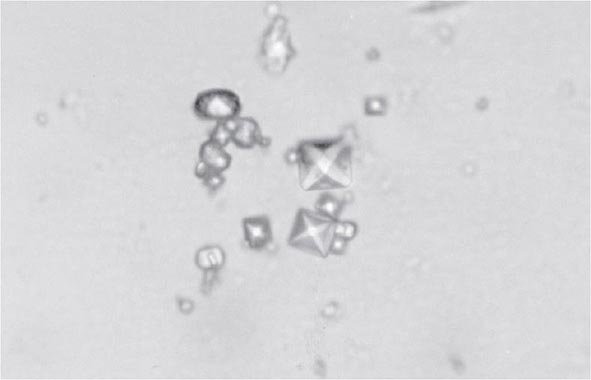
FIGURE 395-4 Bipyramidal and small polymorphic calcium oxalate crystals from synovial fluid are a classic finding in calcium oxalate arthropathy (ordinary light microscopy; 400×).
ACKNOWLEDGMENT
This chapter has been revised for this and the previous two editions from an original version written by Antonio Reginato, MD, in earlier editions of Harrison’s Principles of Internal Medicine.
396 | Fibromyalgia |
DEFINITION
Fibromyalgia (FM) is characterized by chronic widespread musculoskeletal pain and tenderness. Although FM is defined primarily as a pain syndrome, patients also commonly report associated neuropsychological symptoms of fatigue, unrefreshing sleep, cognitive dysfunction, anxiety, and depression. Patients with FM have an increased prevalence of other syndromes associated with pain and fatigue, including chronic fatigue syndrome (Chap. 464e), temporomandibular disorder, chronic headaches, irritable bowel syndrome, interstitial cystitis/painful bladder syndrome, and other pelvic pain syndromes. Available evidence implicates the central nervous system as key to maintaining pain and other core symptoms of FM and related conditions. The presence of FM is associated with substantial negative consequences for physical and social functioning.
EPIDEMIOLOGY
![]() In clinical settings, a diagnosis of FM is made in ∼2% of the population and is far more common in women than in men, with a ratio of ∼9:1. However, in population-based survey studies worldwide, the prevalence rate is ∼2–5%, with a female-to-male ratio of only 2–3:1 and with some variability depending on the method of ascertainment. The prevalence data are similar across socioeconomic classes. Cultural factors may play a role in determining whether patients with FM symptoms seek medical attention; however, even in cultures in which secondary gain is not expected to play a significant role, the prevalence of FM remains in this range.
In clinical settings, a diagnosis of FM is made in ∼2% of the population and is far more common in women than in men, with a ratio of ∼9:1. However, in population-based survey studies worldwide, the prevalence rate is ∼2–5%, with a female-to-male ratio of only 2–3:1 and with some variability depending on the method of ascertainment. The prevalence data are similar across socioeconomic classes. Cultural factors may play a role in determining whether patients with FM symptoms seek medical attention; however, even in cultures in which secondary gain is not expected to play a significant role, the prevalence of FM remains in this range.
CLINICAL MANIFESTATIONS
Pain and Tenderness At presentation, patients with FM most commonly report “pain all over.” These patients have pain that is typically both above and below the waist on both sides of the body and involves the axial skeleton (neck, back, or chest). The pain attributable to FM is poorly localized, difficult to ignore, severe in its intensity, and associated with a reduced functional capacity. For a diagnosis of FM, pain should have been present most of the day on most days for at least 3 months.
The clinical pain of FM is associated with increased evoked pain sensitivity. In clinical practice, this elevated sensitivity may be determined by a tender-point examination in which the examiner uses the thumbnail to exert pressure of ∼4 kg/m2 (or the amount of pressure leading to blanching of the tip of the thumbnail) on well-defined musculotendinous sites (Fig. 396-1). Previously, the classification criteria of the American College of Rheumatology required that 11 of 18 sites be perceived as painful for a diagnosis of FM. In practice, tenderness is a continuous variable, and strict application of a categorical threshold for diagnostic specifics is not necessary. Newer criteria eliminate the need for tender points and focus instead on clinical symptoms of widespread pain and neuropsychological symptoms. The newer criteria perform well in a clinical setting in comparison to the older, tender-point criteria. However, it appears that when the new criteria are applied to populations, the result is an increase in prevalence of FM and a change in the sex ratio (see “Epidemiology,” earlier).
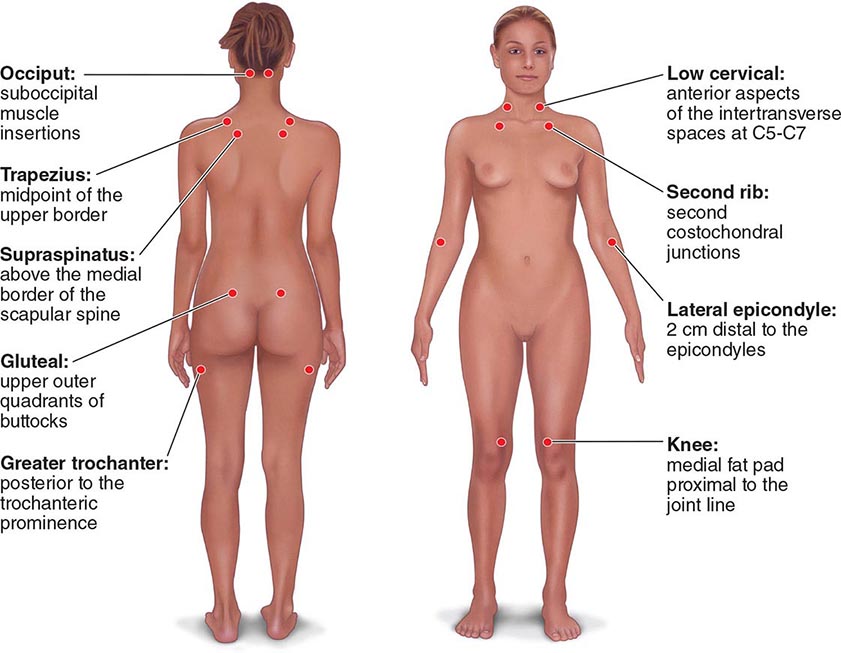
FIGURE 396-1 Tender-point assessment in patients with fibromyalgia. (Figure created using data from F Wolfe et al: Arthritis Care Res 62:600, 2010.)
Patients with FM often have peripheral pain generators that are thought to serve as triggers for the more widespread pain attributed to central nervous system factors. Potential pain generators such as arthritis, bursitis, tendinitis, neuropathies, and other inflammatory or degenerative conditions should be identified by history and physical examination. More subtle pain generators may include joint hypermobility and scoliosis. In addition, patients may have chronic myalgias triggered by infectious, metabolic, or psychiatric conditions that can also serve as triggers for the development of FM. These conditions are often identified in the differential diagnosis of patients with FM, and a major challenge is to distinguish the ongoing activity of a triggering condition from FM that is occurring as a consequence of a comorbid condition and that should itself be treated.
Neuropsychological Symptoms In addition to widespread pain, FM patients typically report fatigue, stiffness, sleep disturbance, cognitive dysfunction, anxiety, and depression. These symptoms are present to varying degrees in most FM patients but are not present in every patient or at all times in a given patient. Relative to pain, such symptoms may, however, have an equal or even greater impact on function and quality of life. Fatigue is highly prevalent in patients under primary care who ultimately are diagnosed with FM. Pain, stiffness, and fatigue often are worsened by exercise or unaccustomed activity (postexertional malaise). The sleep complaints include difficulty falling asleep, difficulty staying asleep, and early-morning awakening. Regardless of the specific complaint, patients awake feeling unrefreshed. Patients with FM may meet criteria for restless legs syndrome and sleep-disordered breathing; frank sleep apnea can also be documented. Cognitive issues are characterized as slowness in processing, difficulties with attention or concentration, problems with word retrieval, and short-term memory loss. Studies have demonstrated altered cognitive function in these domains in patients with FM, though speed of processing is age-appropriate. Symptoms of anxiety and depression are common, and the lifetime prevalence of mood disorders in patients with FM approaches 80%. Although depression is neither necessary nor sufficient for the diagnosis of FM, it is important to screen for major depressive disorders by querying for depressed mood and anhedonia. Analysis of genetic factors that are likely to predispose to FM reveals shared neurobiologic pathways with mood disorders, providing the basis for comorbidity (see later in this chapter).
Overlapping Syndromes Because FM can overlap in presentation with other chronic pain conditions, review of systems often reveals headaches, facial/jaw pain, regional myofascial pain particularly involving the neck or back, and arthritis. Visceral pain involving the gastrointestinal tract, bladder, and pelvic or perineal region is often present as well. Patients may or may not meet defined criteria for specific syndromes. It is important for patients to understand that shared pathways may mediate symptoms and that treatment strategies effective for one condition may help with global symptom management.
Comorbid Conditions FM is often comorbid with chronic musculoskeletal, infectious, metabolic, or psychiatric conditions. Whereas FM affects only 2–5% of the general population, it occurs in 20% or more of patients with degenerative or inflammatory rheumatic disorders, likely because these conditions serve as peripheral pain generators to alter central pain-processing pathways. Similarly, chronic infectious, metabolic, or psychiatric diseases associated with musculoskeletal pain can mimic FM and/or serve as a trigger for the development of FM. It is particularly important for clinicians to be sensitive to pain management of these comorbid conditions so that when FM emerges—characterized by pain outside the boundaries of what could reasonably be explained by the triggering condition, development of neuropsychological symptoms, or tenderness on physical examination—treatment of central pain processes will be undertaken as opposed to a continued focus on treatment of peripheral or inflammatory causes of pain.
Psychosocial Considerations Symptoms of FM often have their onset and are exacerbated during periods of high-level real or perceived stress. This pattern may reflect an interaction among central stress physiology, vigilance or anxiety, and central pain-processing pathways. An understanding of current psychosocial stressors will aid in patient management, as many factors that exacerbate symptoms cannot be addressed by pharmacologic approaches. Furthermore, there is a high prevalence of exposure to previous interpersonal and other forms of violence in patients with FM and related conditions. If posttraumatic stress disorder is an issue, the clinician should be aware of it and consider treatment options.
Functional Impairment It is crucial to evaluate the impact of FM symptoms on function and role fulfillment. In defining the success of a management strategy, improved function is a key measure. Functional assessment should include physical, mental, and social domains. A recognition of the ways in which role functioning falls short will be helpful in the establishment of treatment goals.
DIFFERENTIAL DIAGNOSIS
Because musculoskeletal pain is such a common complaint, the differential diagnosis of FM is broad. Table 396-1 lists some of the more common conditions that should be considered. Patients with inflammatory causes for widespread pain should be identifiable on the basis of specific history, physical findings, and laboratory or radiographic tests.
COMMON CONDITIONS IN THE DIFFERENTIAL DIAGNOSIS OF FIBROMYALGIA |
Inflammatory
Polymyalgia rheumatica
Inflammatory arthritis: rheumatoid arthritis, spondyloarthritides
Connective tissue diseases: systemic lupus erythematosus, Sjögren’s syndrome
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


