Figure 76-1. Schematic illustration of the location of the superior (A) and inferior (B) parathyroid glands from 503 autopsy studies. The more common locations are indicated by the shaded areas. The numbers represent the percentage of glands found at each location. Typically, the glands were posterolateral to the thyroid and above or below the junction of the inferior thyroid artery with the recurrent laryngeal nerve.
Calcium is absorbed in its inorganic form from the duodenum and proximal jejunum. The rate of absorption is precisely regulated according to body calcium status. The calcium in the extracellular fluid is constantly being exchanged with that in the intracellular fluid, the exchangeable bone, and the glomerular filtrate. Calcium reabsorption by the kidney is closely related to that of sodium, and about 99% of the filtered load is reabsorbed under normal conditions.

Figure 76-2. A: Pharyngeal arches in a 5-week embryo. The corresponding pouches extend from within the pharynx into each arch. B: Schematic representation of the differentiating epithelium of the respective pharyngeal pouches.

Figure 76-3. A normal adult parathyroid is composed of about half parenchyma and half fat (×150).
Phosphate
Phosphate anion is also an integral component of most biologic systems. It is critical to the pathways of glycolysis and is the functional group for a number of high-energy transfer compounds, including adenosine triphosphate. It is also the major anion in crystalline bone. Normal levels of plasma phosphate range from 2.5 to 4.3 mg/dL, and the level varies inversely with the serum level of calcium. The relation is such that the product of plasma calcium and phosphate is relatively constant ranging between 30 and 40 (with calcium and phosphate both expressed in mg/dL). When the product is above this level, the potential for the precipitation of calcium phosphate in body tissues increases.
In contrast to the percentage of calcium absorbed, the percentage of phosphate absorbed from the diet is relatively constant, and excretion usually provides the major mechanisms for regulating phosphate balance (Fig. 76-4). Unlike stores of calcium, the readily exchangeable soft tissue stores of phosphate, such as those in muscle, are large.

Figure 76-4. Average daily calcium and phosphate turnover in humans.
Regulation of Calcium and Phosphate Metabolism
The maintenance of calcium and phosphate homeostasis depends on major contributions from three organ systems – the gastrointestinal tract, the skeleton, and the kidneys – with minor contributions from the skin and liver.2 The primary hormonal regulators of this metabolism are PTH, vitamin D, and calcitonin. The actions of each of these hormones in the organs are summarized in Table 76-1.
Parathyroid Hormone
2 PTH is the single most important hormonal regulator of calcium and phosphate metabolism in humans. It has direct effects on the skeleton and kidney and indirect effects on the intestine, mediated through vitamin D. In target tissues, PTH binds first to membrane receptors, activating adenyl cyclase to generate cyclic adenosine monophosphate (cAMP), which regulates other intracellular enzymes.
In bone, the effects of PTH are complex, stimulating both resorption and the formation of new bone. However, sustained elevations of PTH stimulate osteoclasts and inhibit osteoblasts. Osteocytes, in the matrix of cortical bone, may also act to reabsorb matrix in response to PTH, a process referred to as osteocytic osteolysis. Calcium and phosphate mobilization in response to PTH occurs in two phases. Initially, mineral is mobilized from areas of rapid equilibrium. This phase is followed by a more sustained release mediated by newly synthesized lysosomal and hydrolytic enzymes. In the kidney, PTH increases the reabsorption of extracellular fluid calcium at any given concentration, although excess secretion, because of hypercalcemia, increases the net daily amount of urinary calcium excretion. Reabsorption in the proximal tubule and loop of Henle is linked with sodium transport such that factors that alter sodium transport concomitantly alter calcium reabsorption. In contrast, reabsorption in the distal nephron is independent of sodium and directly influenced by PTH. PTH also increases phosphate excretion. This action is accompanied by enhanced bicarbonate secretion. PTH probably has only indirect effects on the gastrointestinal tract, by stimulating the hydroxylation of 25-hydroxyvitamin D to 1,25-dihydroxyvitamin D in the kidney.
Table 76-1 Hormonal Regulation of Calcium and Phosphate Metabolism


Figure 76-5. The parathyroid gland produces a precursor of PTH, prepro-PTH, that is sequentially cleaved to pro-PTH and PTH. PTH secretion is controlled by the extracellular fluid calcium concentration.
PTH is synthesized initially as a precursor, preproPTH, which is sequentially cleaved in the parathyroid gland to proPTH and then to PTH (Fig. 76-5). Secretion of this 84-amino-acid molecule is controlled by a negative feedback loop with extracellular fluid calcium. Most PTH is secreted in this form and then cleaved in the liver into N- and C-terminal fragments. The N-terminus contains most of the biologic activity and is rapidly degraded by the liver, whereas the inactive C-terminus is slowly metabolized by the kidney.
Vitamin D
Vitamin D acts at two major sites. It increases intestinal absorption of calcium and phosphate. In addition, in the skeleton, it promotes mineralization and enhances PTH-mediated mobilization of calcium and phosphate. It has no known direct effect on the kidney.
Vitamin D3, or cholecalciferol, is produced normally by the action of sunlight on 7-dehydrocholesterol in the skin (Fig. 76-6). It is then hydroxylated in the liver (25 position) and kidney (1 position) to form the active 1,25-dihydroxyvitamin D3 (calcitriol). Vitamin D2 is normally present in yeast and fungi but not in humans. It is the major pharmacologic source of vitamin D. Pharmaceutical preparations include vitamin D2 (ergocalciferol), 25-hydroxycholecalciferol (calcifediol), and 1,25-dihydroxycholecalciferol (calcitriol). 1-Hydroxycholecalciferol and dihydrotachysterol are synthetic preparations that require only 25-hydroxylation for activity and so are useful for supplementation in patients with renal failure, who lack the 1-hydroxylase.
Calcitonin
Calcitonin is a 32-amino-acid protein produced by the parafollicular C (calcitonin) cells of the thyroid. The C cells are embryologically derived from the neural crest and, in lower animals, are found in the ultimobranchial bodies, which are glandular structures derived from the lowest branchial pouch. In humans, these structures are incorporated into the superior and lateral aspects of the thyroid lobes.
Total thyroidectomy, with removal of all the C cells, is well tolerated. Calcitonin is not essential for the normal control of calcium metabolism in adult humans. It does inhibit bone resorption and can produce hypocalcemia in experimental animals. It also increases urinary calcium and phosphate excretion. These effects are mediated primarily through cAMP. Several secretagogues for calcitonin have been identified, including catecholamines, gastrin, and cholecystokinin, but the most potent appear to be calcium and pentagastrin. Exogenously administered calcitonin can be useful pharmacologically to reduce serum calcium levels.
Mineral Homeostasis
Under normal conditions, serum calcium and phosphate levels vary minimally during the course of the day. Regulation occurs primarily through PTH but also through a series of feedback loops involving vitamin D and calcitonin (Fig. 76-7). A fall in serum-ionized calcium increases PTH secretion and stimulates the production of 1,25-dihydroxyvitamin D3. Conversely, increases in serum calcium inhibit PTH secretion and the formation of active calciferol.
Pathophysiology
Diseases of the parathyroid glands present almost exclusively as disorders of calcium metabolism. Hypercalcemia is the most common manifestation, and in the patient who presents with an elevated serum calcium level, the differential diagnosis can be complex. A thorough understanding of both hypercalcemia and hypocalcemia is essential for the successful treatment of patients undergoing parathyroid surgery. Primary disorders of plasma phosphate are not usually related to surgical disease and are not discussed in detail here.
HYPERCALCEMIA
Hypercalcemia is a relatively common clinical problem.3,4 In the general population and in hospital outpatients, the incidence is between 0.1% and 0.5%. Most patients in this group have primary hyperparathyroidism. In contrast, hypercalcemia is identified in almost 5% of hospitalized patients, and nearly two-thirds of them have a malignancy.
Clinical Manifestations
The symptoms of hypercalcemia are varied and nonspecific (Table 76-2). Severity is a function of both the magnitude and rapidity of onset of the hypercalcemia. Many of the manifestations are subtle and are evident only in retrospect, after the patient has been successfully treated for the cause of the elevated calcium. Specific symptoms and diagnostic tests are addressed in more detail in the section on hyperparathyroidism.

Figure 76-6. Schematic illustration of the synthesis of vitamin D3. Ergosterol, 1-alpha-hydroxyvitamin D3, and dihydrotachysterol are synthetic preparations of vitamin D.
Differential Diagnosis
Although the diagnosis of primary hyperparathyroidism can, after appropriate investigation, be established with confidence in most patients, all causes of hypercalcemia must be considered and excluded. The multiple causes of hypercalcemia are listed in Table 76-3.
Etiology
Hyperparathyroidism
The diagnosis of hyperparathyroidism is discussed in detail later. Patients typically have elevated plasma concentrations of calcium and PTH, increased urinary excretion of calcium, and a low plasma concentration of phosphate.
Malignancy
Generally, patients with hypercalcemia and malignancy (humoral hypercalcemia of malignancy) can be classified into two groups.5–7 Patients with solid tumors, such as lung carcinoma (25% of all cases of humoral hypercalcemia of malignancy); breast carcinoma (20%); squamous cell carcinoma of the head, neck, esophagus, or female genital tract (19%); or renal cell cancer (8%), account for three-fourths of all cases. Humoral hypercalcemia of malignancy in this setting generally appears late in the disease, with nearly all patients having known, or readily evident, malignancy. These patients have elevated levels of serum calcium, low levels of serum phosphorus, and elevated levels of urinary cAMP, consistent with increased PTH activity but normal or low-serum PTH levels. The hypercalcemia is now known to be caused by PTH-related protein secreted by the tumor, rather than by the bony metastases that many of these patients have because of the advanced nature of their cancers. In the second group, accounting for one-fourth of cases, are patients with hematologic malignancies, such as multiple myeloma, certain lymphomas and leukemias, and a subset of the patients with breast cancer. These patients have elevated levels of serum calcium, but in contrast to most patients with solid tumors and humoral hypercalcemia of malignancy, they have elevated levels of serum phosphate and low levels of urinary cAMP. These patients always have lytic bony lesions and histologically demonstrate increased osteoclast bone resorption adjacent to tumor cells. This osteoclast-activating activity is an effect of cytokines, mainly interleukin-1 beta and tumor necrosis factor-beta (lymphotoxin). These cytokines promote local net bone resorption and thus produce hypercalcemia and hyperphosphatemia.

Figure 76-7. Feedback loops involved in the regulation of serum calcium and phosphorus. PTH, parathyroid hormone; CT, calcitonin.
DIAGNOSIS
Table 76-2 Clinical Features of Hypercalcemia

Vitamin D and Vitamin A Intoxication
When administered in excess, vitamins A and D can produce hypercalcemia. Affected patients tend to have normal or elevated serum phosphate levels associated with a low PTH level. Metastatic calcification may occur.
Thiazide Diuretics
Thiazides may increase serum calcium levels to a mild degree, primarily through hemoconcentration and decreased renal excretion. Serum phosphate may also be depressed. It often takes several weeks for the hypercalcemia to resolve after the medication is discontinued.
Hyperthyroidism
Hyperthyroidism is associated with increased bone resorption. Often, the plasma PTH is low, and a history of other thyrotoxic symptoms can be elicited. The hypercalcemia usually resolves as the patient becomes euthyroid.
Milk–Alkali Syndrome
Typically, the milk–alkali syndrome occurs in patients with peptic ulcers who consume large quantities of milk and absorbable antacids. Usually, some degree of renal failure is present. PTH levels are low. This syndrome has become much less common with the increased use of nonabsorbable antacids, histamine 2-receptor antagonists, and proton pump inhibitors as therapy for peptic ulcer disease.
ETIOLOGY
Table 76-3 Causes of Hypercalcemia

Sarcoidosis and Other Granulomatous Diseases
These syndromes are associated with hypersensitivity to vitamin D. The granulomas can convert inactive vitamin D to its active form. Patients have elevated plasma globulins and low PTH levels. The administration of large doses of corticosteroids for 10 days usually reduces the hypercalcemia. Biopsy of lymph nodes or the liver may confirm the diagnosis.
Familial Hypocalciuric Hypercalcemia
This disease is an asymptomatic, autosomal dominant condition characterized by mild to moderate hypercalcemia, hypocalciuria, and normal or only slightly elevated PTH levels. It develops in people heterozygous for a mutation in the calcium-sensing receptor.8–10 The mutation causes an increase in the set point for extracellular calcium concentration, so that the “normal” calcium level is higher in these people than in the normal population. No treatment is necessary, although people with this disease should receive genetic counseling. Neonatal severe hyperparathyroidism, which can be fatal, develops in children homozygous for mutations in this receptor. Treatment for neonates with this disease is controversial, but they appear to benefit from early surgical management.
Immobilization
Immobilization produces hypercalcemia by increasing the ratio of bone resorption to bone formation. These patients can usually be distinguished by history, although on laboratory evaluation they have elevated serum levels of calcium and phosphate and a decreased serum concentration of PTH. Often, hypercalciuria is present, which may lead to the development of renal stones. Treatment is early mobilization and forced diuresis.
Other Causes
A variety of other diseases may produce hypercalcemia. For example, Paget disease (osteitis deformans) typically causes mild elevations in serum calcium. Paget disease can be diagnosed on the basis of the characteristic radiographic lesion. Adrenal insufficiency may be associated with hypercalcemia, although the symptoms are typically those of the primary abnormality. Lithium therapy appears to produce hypercalcemia by altering the parathyroid set point for inhibition by calcium, and, over long courses of therapy, may also be associated with hyperparathyroidism. Idiopathic hypercalcemia of infancy is a rare disorder that is probably the result of hypersensitivity to vitamin D. It occurs in infants with mental retardation and is satisfactorily treated with glucocorticoids. Other causes include aluminum-induced renal osteomalacia and a host of analytic errors related to improper specimen collection with prolonged tourniquet times, tube contamination, and instrument drift.
Medical Treatment
Although the choice of therapy is tailored to the cause of the hypercalcemia, several general measures can prove effective.11,12
For the patient with mild hypercalcemia, a trial of a decrease in dietary calcium is indicated. A reduction in intake of milk and other dairy products is suggested, along with discontinuation of thiazide diuretics and vitamin D preparations. Mobilization prevents bone demineralization.
Patients with more marked hypercalcemia or severe symptoms should be admitted to the hospital for treatment, with careful observation and monitoring. In the patient with severe hyperparathyroidism, although the definitive therapy is surgical, it is unwise to proceed with neck exploration until the calcium has been reduced to near-normal levels. The mainstay of therapy is intravenous hydration, preferably with normal saline solution in sufficient quantities to maintain the urine output above 100 mL/hr. These patients are often dehydrated before therapy, and fluid can be administered intravenously at a rate of 200 mL/hr. Caution must be exercised in older patients, whose cardiac reserve may be marginal. This therapy exploits the parallel handling of calcium and sodium by the kidneys. The diuretic furosemide also increases sodium and calcium excretion but should not be used until the patient is well hydrated.
The end points of therapy are a decrease in the serum calcium level and a reduction of symptoms. Diuresis with saline solution is usually effective when the hypercalcemia results from hyperparathyroidism or a benign cause. In contrast, the hypercalcemia of malignancy may produce severe symptoms associated with extremely high serum calcium levels that are difficult to control. In this setting, a variety of other measures may be considered (Table 76-4). Some of the agents used to treat hypercalcemia cause significant toxicity, and close patient monitoring is required during treatment. Calcitonin is a fairly weak hypocalcemic agent, but it acts rapidly and is associated with less toxicity than many of the other drugs.12 Salmon calcitonin is the most potent preparation. Treatment with glucocorticoids is particularly efficacious in patients with sarcoidosis and other granulomatous diseases. Plicamycin has proved useful in patients with hypercalcemia of malignancy, but it causes a cumulative toxicity (thrombocytopenia, hepatotoxicity, and nephrotoxicity). Bisphosphonates inhibit osteoclast activity directly. These agents are administered orally or parenterally and are particularly efficacious, although long-term use may be associated with significant osteomalacia.7 Prostaglandin synthetase inhibitors were initially considered useful, but their efficacy has proved to be limited. Intravenous phosphates and chelating agents have largely been abandoned because of their severe toxicity; however, oral phosphates may be beneficial in patients requiring prolonged therapy.
HYPOCALCEMIA
Hypocalcemia can occur as a consequence of various acquired and hereditary diseases.13 Generally, these disorders produce a deficiency or defect in the action of either PTH or vitamin D. It is most commonly a significant clinical problem after neck operation for thyroid disease. Chronic vitamin D deficiency is associated with compensatory PTH excess. The end result is rickets in children or osteomalacia in adults.
TREATMENT
Table 76-4 Treatment of Hypercalcemia

Clinical Features
The major signs and symptoms of hypocalcemia are a direct consequence of the reduction in plasma levels of ionized calcium, which increases neuromuscular excitability (Table 76-5). The earliest clinical manifestations are numbness and tingling in the circumoral area, fingers, and toes. Mental symptoms are also common. Patients become anxious, depressed, and occasionally confused. Tetany may develop, characterized by carpopedal spasm, tonic–clonic convulsions, and laryngeal stridor. The magnitude of symptoms at any given plasma concentration of ionized calcium varies from patient to patient. On physical examination, contraction of the facial muscles is elicited by tapping anterior to the ear, over the facial nerve (Chvostek sign), although this sign may be present in 10% of normocalcemic patients. Trousseau sign is elicited by occluding blood flow to the forearm for 3 minutes. The development of carpal spasm indicates hypocalcemia, although the test is unpleasant and clinically impractical.
Etiology
Some of the causes of hypocalcemia are listed in Table 76-6. The most common cause of hypocalcemia by far is excision of or damage to the parathyroid glands during thyroid surgery.
Postoperative Hypoparathyroidism
Postoperative hypoparathyroidism commonly develops after total thyroidectomy.13,14 Most patients undergoing operation on the thyroid experience some alteration in serum calcium, although they often are asymptomatic; the low calcium probably represents contusion or temporary alteration of the blood supply to the parathyroid glands. The hypocalcemia is usually transient and is not treated unless significant symptoms develop. Occasionally, in hyperparathyroid patients who have parathyroidectomy and significant bone disease, a marked skeletal deposition of calcium and symptomatic hypocalcemia occurs, the so-called bone hunger. The plasma calcium usually reaches its nadir at 48 to 72 hours after surgery and then slowly returns to normal within several days. These patients may require calcium and vitamin D therapy for weeks or months after parathyroidectomy.
DIAGNOSIS
Table 76-5 Clinical Features of Hypocalcemia

Idiopathic Hypoparathyroidism
A less common form of hypoparathyroidism is idiopathic lack of function. It occurs both sporadically and in families. In some cases, it develops as part of a polyglandular disorder and is thought to have an autoimmune basis. DiGeorge syndrome is a group of congenital disorders involving the branchial pouches that produce partial or complete agenesis of the thymus and parathyroid glands. Hypoparathyroidism can also develop in newborns as a result of prenatal suppression of the fetal parathyroid glands as a consequence of maternal hypercalcemia.15 It is also common in otherwise normal but premature infants.
ETIOLOGY
Table 76-6 Causes of Hypocalcemia

Vitamin D Deficiency
Vitamin D deficiency can occur as a result of dietary deficiency or lack of sun exposure. Likewise, renal disease produces a decrease in the 1-hydroxylase activity necessary for the formation of active vitamin D. The result is a decrease in calcium absorption and an increased secretion of PTH by the stimulated parathyroid glands. Osteomalacia, abnormal fractures, and the deformities of rickets may result.
Pseudohypoparathyroidism
Pseudohypoparathyroidism is a familial disease characterized by a rotund appearance, shortening of the extremities, and sometimes mental deficiency. The defect is not in PTH secretion; in fact, most patients have elevated plasma levels of PTH with evidence of increased bone resorption. Rather, the kidney is unresponsive to the hormone, and as a consequence, hypocalcemia and hyperphosphatemia develop. The deficit appears to be in the renal adenyl cyclase system.
Hypomagnesemia
This unusual deficit may result from chronic alcoholism, malabsorption, parenteral nutrition, or increased renal clearance during therapy with aminoglycosides. The deficit appears to block the physical response to PTH in addition to its release from the parathyroid gland.
Other Causes
In short-gut syndrome, after extensive small-bowel resection or bypass, or after some forms of bariatric surgery, vitamin D and calcium may be absorbed in insufficient quantities. In pancreatitis, the massive soft tissue destruction and saponification that occur with hemorrhagic disease may sequester significant amounts of calcium in the retroperitoneum. Some undefined systemic factor also appears to contribute to hypocalcemia in these patients. Hypoalbuminemia causes a reduction in the total plasma calcium level, although the level of ionized calcium remains within the normal range and patients are asymptomatic. Circulatory substances, such as the citrate used to anticoagulate banked blood and radiographic contrast media, may bind to calcium. In patients with osteoblastic metastases, particularly associated with prostate carcinoma, hypocalcemia has been attributed to increased calcium flux into the lesions. Toxic shock syndrome is sometimes associated with hypocalcemia, but the mechanism has not been defined. Acute hyperphosphatemia, as a consequence of exogenous administration of phosphate or during the cytolytic chemotherapy of highly responsive tumors (e.g., Burkitt lymphoma and acute lymphoblastic leukemia), may produce symptomatic hypocalcemia associated with soft tissue calcification.
Treatment
The treatment of hypocalcemia is summarized in Table 76-7. For acute symptomatic hypocalcemia, calcium should be administered intravenously. Calcium gluconate is less irritating to the veins than calcium chloride, and the calcium release is slower, without a risk for overcorrection. Usually, 20 to 30 mL of 10% solution is infused over a 15- to 20-minute period, and then 50 to 100 mL is administered over the next 12 hours in adults. A practical guide after an initial bolus dose includes 60 mL of 10% calcium gluconate in a 500-mL bag of dextrose 5% in water, infused at 1 mL/kg/hr, and adjusted every 4 hours based on the serum level of calcium and patient symptoms. Bicarbonate precipitates any calcium infused through the same intravenous line. Serum magnesium should always be measured, and hypomagnesemia should be corrected if present. In patients with convulsions from advanced tetany, diphenylhydantoin therapy is useful, but symptoms should never be allowed to progress to this point.
TREATMENT
Table 76-7 Treatment of Hypocalcemia

Long-term therapy is gauged on the basis of symptoms. In the postoperative patient, the continued stimulus of mild hypocalcemia to any remaining parathyroid gland tissue may prove useful. Concomitant therapy with calcium and vitamin D is effective in a timely fashion. A starting dose of 2 g of oral calcium carbonate per day in divided doses is usually well tolerated. Vitamin D can be administered as calcitriol, an active synthetic vitamin D analog. Most adults respond to a dose of 0.5 to 1.0 mcg/day; reduced doses may be necessary for patients with renal dysfunction.
HYPERPARATHYROIDISM
Definitions
Parathyroid neoplasms are rarely identified by physical enlargement but rather are sought because of the peripheral effects of excess hormone. Primary hyperparathyroidism develops spontaneously, without apparent cause but possibly in response to exogenous stimuli. When the normal control of serum calcium is disturbed and the autonomous production of PTH is increased, the state is referred to as primary hyperparathyroidism. This category includes both benign single- and multiple-gland enlargements and the much rarer parathyroid carcinoma. In some cases, the disease is familial. In contrast, secondary hyperparathyroidism occurs when a defect in mineral homeostasis leads to a compensatory increase in parathyroid function. This occurs most commonly in response to renal disease but may also develop as a consequence of the hypocalcemia associated with some diseases of the gastrointestinal tract, bones, or other endocrine organs. Occasionally, with prolonged secondary stimulation, the hyperfunctioning glands are no longer physiologically responsive to an increase in ionized calcium. This uncommon (affecting about 2% of patients after renal transplantation), relatively autonomous state referred to as tertiary hyperparathyroidism, develops most commonly after renal transplantation when the renal defect in calcium homeostasis is corrected.
Incidence
The advent in the 1970s of the widespread assessment of serum calcium as part of automated multichannel analysis has considerably altered our understanding of hyperparathyroidism. Before that time, primary hyperparathyroidism was thought to be a relatively rare condition. Most patients presented with symptoms of disease, usually renal stones or bony manifestations. Currently, most patients are asymptomatic or have only vague symptoms or signs that can be related to hyperparathyroidism.16,17 Occasionally, patients recognize that they had symptoms only after their well-being improves following parathyroidectomy. Incidence varies with both age and gender (Table 76-8), but hyperparathyroidism is believed to develop in about 50 to 100 people per 100,000 in the general population, with approximately 50,000 new cases occurring annually in the United States.18 Marked variations have been noted worldwide; the reasons for these differences remain unclear.
Table 76-8 Age-Specific and Gender-Specific Incidence of Primary Hyperparathyroidism

Etiology
The cause of primary hyperparathyroidism is not known. Although the sequence of progression from secondary to tertiary disease in response to chronic stimulation has a logical appeal, it is difficult to draw parallels with primary disease. Most patients with primary hyperparathyroidism have disease of a single rather than of multiple glands, which is not what might be predicted if an external stimulus were part of the pathophysiology. Hyperparathyroidism is most common in postmenopausal women, the population group with the highest incidence of osteoporosis and the most significant alterations in calcium and phosphate metabolism. Loss of renal function with aging is associated with elevations in PTH and decreases in phosphate clearance. It has been suggested but not demonstrated that a renal calcium leak, if sufficient, might result in a chronic calcium deficit stimulating the parathyroid glands.
Genetic studies of parathyroid adenomas have described an oncogene (PRAD1) that may be one step in the path to neoplasia in these tumors. Overexpression of the normal PRAD1 gene, also known as cyclin D1, allows progression of the cell cycle from the G1 phase to the S phase, thus promoting cellular growth and division. PRAD1 is overexpressed in 20% to 40% of parathyroid adenomas; further research may reveal other genetic alterations that contribute to the neoplastic growth. The MEN1 tumor-suppressor gene has also been implicated in the molecular pathogenesis of sporadic hyperparathyroidism. About 15% to 20% of sporadic parathyroid adenomas have either somatic mutation or biallelic deletion of the MEN1 gene.19,20
Hyperparathyroidism occurs in several familial forms. It is a major component of the multiple endocrine neoplasia (MEN) syndromes types 1 and 2A. The parathyroid disease of MEN-1 syndrome is multiple parathyroid adenomas that appear with increasing frequency over the patient’s lifetime.21 In other families, hyperparathyroidism is inherited in an autosomal dominant fashion without other manifestations of MEN-1 or MEN-2; some have osseous abnormalities (tumor–jaw syndrome) and some apparently have isolated disease.

Figure 76-8. Parathyroid adenoma. The tumor consists of sheets of neoplastic chief cells and is separated from normal parenchyma by a thin capsule.
Pathology
Single-Gland Versus Multiple-Gland Disease
Microscopically, the cell most commonly involved in primary hyperparathyroidism is the chief cell. Less frequently, the oxyphil cell is the predominant cell type. Diseased glands typically have an increase in the proportion of stromal cells and a reduction in the proportion of stromal fat. Single diseased glands, or adenomas, have been classically described with a predominance of chief cells centering in a single focus, with a compressed rim of surrounding normal tissue (Fig. 76-8). In contrast, parathyroid hyperplasia has been characterized as a diffuse proliferation of clear cells in multiple glands, with little remaining normal tissue (Fig. 76-9). These criteria have proved totally unreliable. Although pathologic studies can usually distinguish parathyroid glands from other tissue, they may not prove useful beyond this capacity. Intraoperative decisions frequently depend on recognizing disease of one or more parathyroid glands, and in this regard, the histologic description of adenoma or hyperplasia is generally unreliable in primary hyperparathyroidism.22
Patients with multiple-gland disease may have one gland that appears to be an adenoma and another that appears diffusely involved or even histologically normal with gross enlargement. The most reliable index of abnormality is the determination of glandular enlargement by visual inspection. The incidence of single- and multiple-gland enlargement as judged by visual inspection in 66 consecutive patients with hyperparathyroidism is shown in Table 76-9. The visual assessment and judgment of the experienced surgeon have proved to be an effective basis for intraoperative decisions. This approach requires that all four parathyroid glands be evaluated at the time of operation. However, the recent ability to rapidly measure PTH during operation has provided a method to assess gland function rather than size. This experience indicates that there may be more enlarged glands than there are hyperfunctioning glands.23
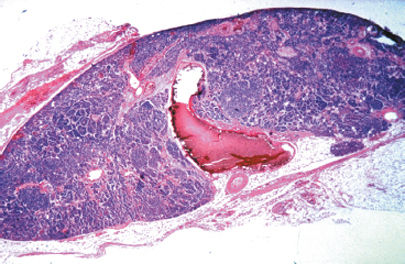
Figure 76-9. Primary parathyroid hyperplasia. The normal adipose tissue of the gland has been replaced by sheets and trabeculae of hyperplastic chief cells.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


