Changes in oxygen (O2) tension are detected by O2-sensitive K+ and Ca2+ channels, and a suprathreshold increase of cytosolic Ca2+ triggers neurotransmitter (i.e., dopamine) release, which activates afferent nerve terminals (13). Despite the immense amount of experimental research on carotid bodies in animals, the precise mechanism(s) of O2 sensing and the basic chemosensor(s) or transducer(s) is still not entirely clear. Numerous proposals have been put forth. Proposed candidates include reactive oxygen species, mitochondria, AMP-activated kinase, hemoxygenase-2, and succinate dehydrogenase (SDH). No single mechanism has been shown to explain the multitude of accumulated data, and it may be that a cascade involving several mechanisms is required to accomplish the necessary homeostatic response to arterial hypoxemia (14–16). It has been possible to study dissociated chemoreceptor cells or intact carotid body in culture, and in a recent study, chemoreceptor cells were not sensitive to hypoglycemia (17).
MORPHOLOGY AND ANATOMIC DISTRIBUTION
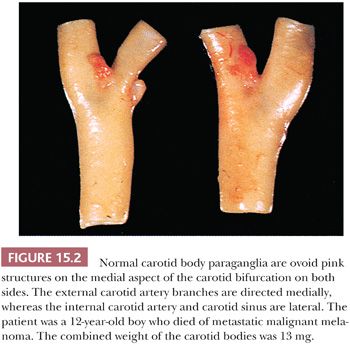
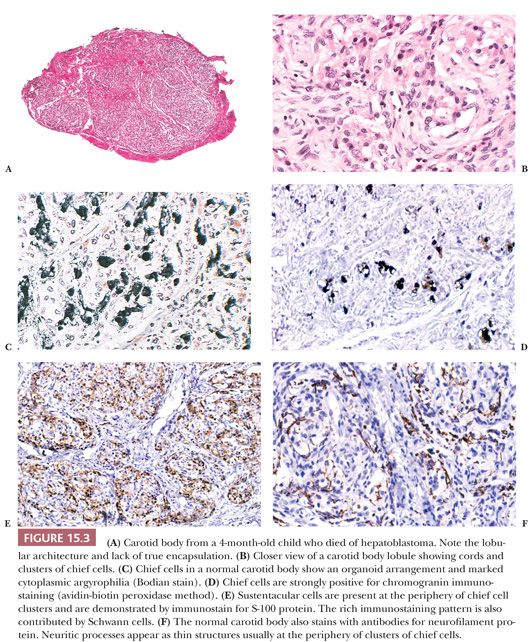
Carotid bodies are the largest compact collection of paraganglia in the head and neck region; they appear as a small ovoid structure on the medial aspect of the carotid bifurcation on each side of the neck (Fig. 15.2). The average combined weight of carotid bodies in adults without chronic hypoxia or systemic hypertension is approximately 12 mg (18,19). Paraganglia located elsewhere in the head and neck region have a nearly identical histomorphologic picture, but they are smaller and usually lack the compact lobular architecture of the carotid bodies (Fig. 15.3A). The basic anatomic unit of the carotid body is the lobule, which contains clusters (“zellballen”) or cords of chief cells (Fig. 15.3B). Within the lobule, a variety of different cells are present, including chief cells and sustentacular or glomus type II cells, as well as pericytes, endothelial cells, and Schwann cells. Chief cells can be vividly depicted by staining for cytoplasmic argyrophilia (Fig. 15.3C) or chromogranin A (Fig. 15.3D), and this organoid arrangement of chief cells is vaguely recapitulated in the neoplasms derived from them. Sustentacular cells are located at the periphery of clusters of chief cells and can be demonstrated by immunostain for S-100 protein (Fig. 15.3E); this immunostain will also highlight the Schwann cells in the normal carotid body and other paraganglia. Sustentacular cells are typically present in paragangliomas at all anatomic sites in the head and neck region and the sympathoadrenal neuroendocrine system. The rich component of neuritic processes (dendritic and/or axonal) can be nicely demonstrated with immunostain for neurofilament protein (Fig. 15.3F).
Carotid body and other paraganglia contain catecholamines, and chief cells have been shown to have enzymes involved in catecholamine synthesis. The immunophenotype of these endocrine cells and tumors derived from them, however, are remarkably diverse. Paraganglia in the head and neck region are located at a variety of sites, including the middle ear, adventitia of the jugular bulb, ganglion nodosum of the vagus nerve, larynx, and base of the heart (Fig. 15.1). This anatomic distribution in some areas parallels the branchial arch derivatives that in aquatic species correspond to gill arches. In some cases, paragangliomas have been reported at sites where normal paraganglionic tissue has not yet been described in humans. Examples include paragangliomas primary in liver (20), ovary (21), bone (22), and parathyroid (23).
HYPERPLASIA OF CHEMORECEPTOR PARAGANGLIA
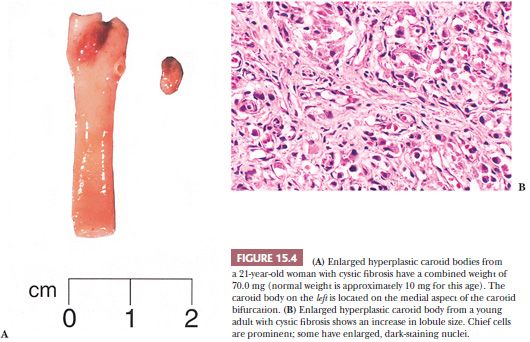
Carotid body enlargement was reported by Arias-Stella (24) in natives born and living in the Peruvian Andes 14,350 feet above sea level. This was confirmed in other studies (25) and also by Saldana et al. (26), who reported a 10-fold increase in incidence of “chemodectomas” at high altitude. An increased incidence of carotid body paragangliomas (CBPs) has been reported in other locations at high altitude (27,28). Carotid body hypertrophy and hyperplasia have also been observed in humans under normobaric conditions, for example, in patients with chronic obstructive pulmonary disease (18,29), systemic hypertension (18), and some cases of cystic fibrosis (Fig. 15.4A,B) and cyanotic congenital heart disease (19,30). Chronic hypoxia leads to hypersensitivity of the carotid bodies with subsequent morphologic and neurochemical changes in the carotid body, including carotid body enlargement, hyperplasia of glomus cells, and neovascularization (31,32). Hyperplasia of vagal paraganglia (33) and aorticopulmonary paraganglia (34) suggests a similar role in chemosensation. Chemoreceptor hyperplasia in most cases is presumably a compensatory response to prolonged and severe hypoxemia (24). Although on rare occasion paragangliomas associated with hypoxemia related to conditions other than high altitude have been reported (35–37), the increased risk of developing a paraganglioma appears to be negligible in patients at sea level, with or without hypoxemia.
PARAGANGLIOMAS OF THE HEAD AND NECK REGION
CAROTID BODY PARAGANGLIOMA
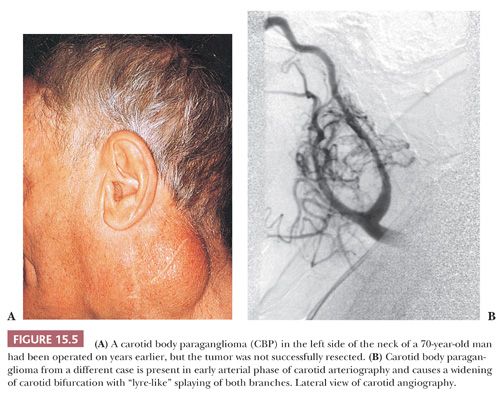
Tumors of this type have been referred to as chemodectomas (chemeia, meaning “infusion”; deschesthai, meaning “to receive”; and oma, meaning “tumor”) (38), and this term has been used synonymously for paragangliomas at other sites in the head and neck region. There is no evidence, however, that any of these tumors have a functional role in chemosensation. The first CBP was reported by Marchand (39) in 1891 in a patient who had been operated on by Riegner in 1880. CBPs typically appear as a slow-growing, painless mass near the angle of the mandible (Fig. 15.5A) (34,40–43). In some patients, there may be signs or symptoms of cranial nerve palsy, such as dysphagia or dysphonia; the carotid sinus syndrome is an unusual feature, with bradycardia and syncopal episodes. On rare occasion, CBPs and other head and neck paragangliomas may be functional, with signs or symptoms of excess catecholamine secretion (34,44,45). The average age at diagnosis is usually in the fifth decade of life, and the average duration of symptoms is approximately 4 years (43,44). Some series report a roughly equal gender distribution, whereas others show a slightly higher incidence in women (46–48). It has been estimated in the sporadic setting that 4% to 8% of patients with CBPs have bilateral tumors (49,50). CBPs are more often bilateral when arising in a familial setting. Occasionally, there may be multiple paragangliomas at other sites, such as the vagus nerve, orbit, middle ear, and base of the heart (51,52). Preoperative selective angiography may be useful for delineating tumor location (Fig. 15.5B), defining blood supply, and providing a route for selective embolization if this is considered before surgery. Preoperative embolization can decrease blood loss in patients with paragangliomas of the head and neck region (48,53) and other sites (54).
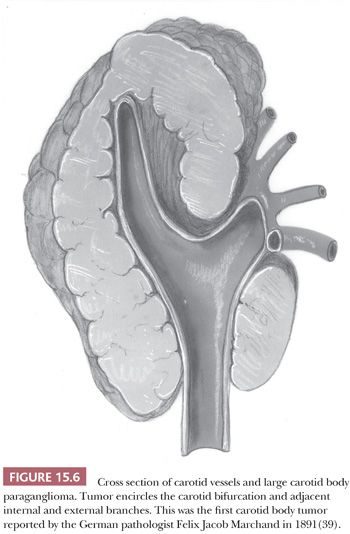
In a large study of CBPs by Shamblin et al. (42), three groups of tumors were identified—group I CBPs which were surgically resectable without significant trauma to the adjacent artery, group II tumors which appeared to partially surround the carotid vessel(s) and were adherent to the adventitia, and group III CBP with a more intimate adherence to the entire circumference of the carotid bifurcation (42). The tumor first reported by Marchand (39) was a group III CBP (Fig. 15.6).
JUGULOTYMPANIC PARAGANGLIOMA
Jugulotympanic paraganglioma (JTP) has a predilection for women, and these tumors arise from microscopic collections of paraganglia, which were described by Guild (55,56) in a study involving serial sectioning of temporal bones; the first case of a patient with JTP was reported by Rosenwasser (57) in 1945. These paraganglia (average of two to three in each temporal bone) may be found along the course of the Jacobsen nerve (tympanic branch of the 9th cranial nerve), the nerve of Arnold (auricular branch of the 10th cranial nerve), the adventitia of the jugular bulb, the osseous canal connecting the jugular fossa to the middle ear cavity, or within the middle ear—usually over the cochlear promontory. The varied locations of these paraganglia and the complex anatomy of this area form the basis for the different clinical presentations of patients with JTP (58). Small tumors arising over the cochlear promontory (tympanic paraganglioma) may arise as an aural polyp, with filling of the middle ear cavity or extension into the external ear canal. JTP can involve the temporal bone, with intracranial extension, or appear as a mass at the base of the skull, with erosion of the jugular foramen. Rarely, these tumors display clinical evidence of malignancy, primarily metastatic disease. The most common site of metastasis includes lymph nodes, skeleton, lungs, and liver (40). The histologic appearance of these tumors is usually not predictive of biologic behavior.
VAGAL PARAGANGLIOMA
These tumors also have a predilection for women in the fourth and fifth decades of life and usually develop as a lateral neck mass with extension up to the base of the skull (43). Vagal paragangliomas (VPs) arise from paraganglia located at the level of or just below the nodose ganglion of the vagus nerve that are usually multiple on either side of the neck (33,59). They have been referred to as vagal body paraganglia, although they are anatomically dispersed microscopic collections of cells. As with CBPs, these tumors may protrude medially with deviation of oropharyngeal structures, such as the palatine tonsil, or extend even higher up into the nasopharynx. They may also demonstrate intracranial extension (60). Vagus nerve palsy may be apparent and may be accompanied by hoarseness and dysphagia (61). VPs arise higher in the neck than CBPs and usually cause anterior bowing of the carotid vessels without direct involvement of the carotid bifurcation. The tumor can also extend into the jugular foramen at the base of the skull (60).
LARYNGEAL PARAGANGLIOMA
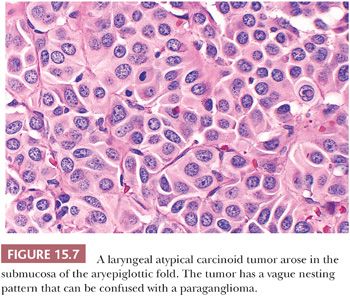
Laryngeal paragangliomas (LPs) arise from microscopic collections of paraganglia distributed on either side of the larynx that form a superior and inferior group (62); these tumors usually evolve from superior laryngeal paraganglia above the anterior part of the vocal cords near the aryepiglottic fold, but they can also develop inferiorly and appear in the subglottic or tracheal area. Hoarseness and dysphagia are the most common complaints (63). In the literature, LPs have been associated with a very high rate of malignancy (64), but most of these “alleged” malignant paragangliomas have undoubtedly been confused with laryngeal atypical carcinoid tumor of the larynx (Fig. 15.7) (65,66). At present, it is believed that true LPs have very limited malignant potential (67,68).
AORTICOPULMONARY PARAGANGLIOMA
Aorticopulmonary paragangliomas (APPs) arise from small collections of paraganglia that are located at various sites, both dorsal and ventral, at the base of the heart in relation to the great vessels (69). Some paraganglia can be found above the aortic arch in relation to the subclavian arteries. APPs can cause symptoms or signs such as hoarseness, dysphagia, chest pain or discomfort, and, rarely, hemoptysis or the superior vena cava syndrome (70). There appears to be a slight predominance among women. The average size of APPs is approximately 7.5 cm (range, 1.2 to 17 cm) (70). Some tumors can involve the pericardium or heart directly (cardiac paragangliomas) (71). A pigmented cardiac paraganglioma has also been reported (72).
OTHER PARAGANGLIOMAS
Paragangliomas have been reported at a variety of other sites in the head and neck region, including the orbit (73–75), nasal cavity (76), hypopharynx (77), parotid gland (78), cheek (79), palatine tonsil (80), pineal (81), and sellar region (82) including Meckel’s cave (83). The thyroid gland is an uncommon but well-recognized primary site, and recently, a sclerosing paraganglioma was reported (84). They have a histologic appearance similar to CBP. Rare examples of primary pulmonary paragangliomas have also been described (85,86). These tumors may arise from intrapulmonary paraganglia, as described by Blessing and Hora (87), although some may be very difficult to distinguish from a bronchial carcinoid tumor. One example has been reported as multicentric metachronous pulmonary and intravagal paragangliomas with an identical immunophenotype; they showed negative results for epithelial markers, as one would expect for paragangliomas (88). A recent case of Cushing syndrome resulting from an adrenocorticotropic hormone (ACTH)–secreting primary pulmonary paraganglioma has been reported (89). A large primary paraganglioma of the lung was reported measuring 13 cm in diameter (90).
GROSS PATHOLOGY
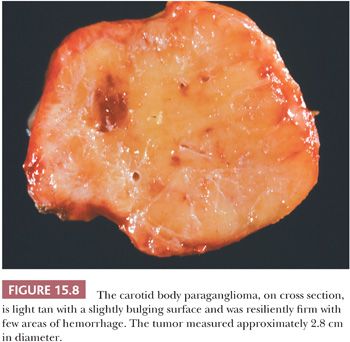
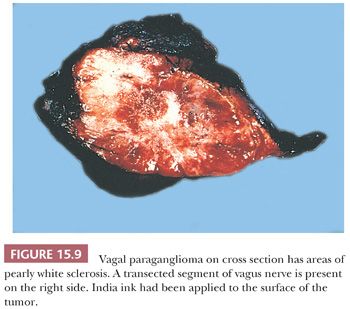
Paragangliomas are rubbery, firm, and usually well demarcated, with expansile borders (Fig. 15.8). The compressed rim of connective tissue surrounding the tumor is best regarded as a fibrous pseudocapsule. Some tumors, such as JTP, may be small and irregular or fragmented, making gross examination difficult. On cross section, these tumors have a meaty to light tan appearance and, if closely scrutinized, may have punctate to linear vascular markings that are often retracted slightly beneath the cut surface. Sometimes, there are intersecting bands of fibrous tissue or intense sclerosis (Fig. 15.9). The average sizes of paragangliomas recorded in one series are as follows: CBPs, 3.8 cm (range, 1.8 to 8.5 cm); VPs, 4.0 cm (range, 2.0 to 6.0 cm); and LP, 2 to 5 cm in diameter (91). The tumor volume was calculated by Nora et al. (92) for CBPs; it ranged from 2.0 to 164 cm3. Larger tumor volume or size correlates with increasing difficulty of surgical resection and longer duration of tumor.
Mechanical manipulation of tumor during surgery may cause a deep red-brown discoloration as a result of congestion and hemorrhage. Areas of degenerative change with necrosis and cystic alteration are very uncommon; however, foci of ischemic necrosis can be seen in some tumors that have been embolized before surgical removal. On rare occasion, a paraganglioma can invade the lumen of the carotid artery or cause total occlusion of a large vessel (93,94).
MICROSCOPIC PATHOLOGY
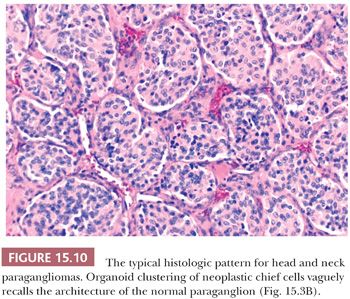
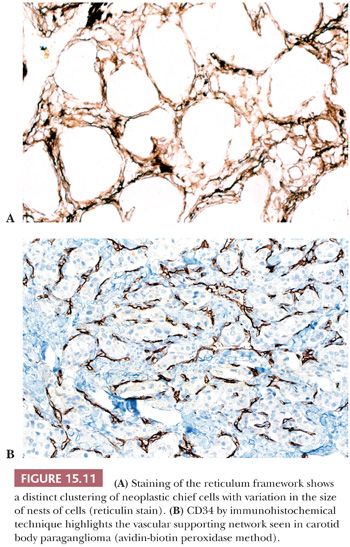
In general, the microscopic patterns encountered in head and neck paragangliomas do not allow one to reliably distinguish between tumors arising at different anatomic sites (34,40). The most typical pattern is a discrete or organoid arrangement of neoplastic chief cells often referred to as zellballen (Fig. 15.10) (95). Several different patterns were recognized by LeCompte (96)—the usual (Fig. 15.10), adenoma-like, and angioma-like. The cell cytoplasm is eosinophilic and faintly granular with indistinct borders. Some tumors may have cells with abundant, deeply eosinophilic cytoplasm, thus imparting an oncocytic appearance (34,40). The orientation of neoplastic chief cells within individual cell clusters tends to be haphazard, without polarization along the fibrovascular septa, and there is no formation of true rosettes or acini, as one might see in other endocrine tumors, such as carcinoids. The nesting pattern can vary in size, and it is most vividly accentuated by staining for reticulum (Fig. 15.11A); the rich vascularity of these tumors can be clearly demonstrated by immunostains for endothelial markers (Fig. 15.11B). There is a network of supporting cells referred to as sustentacular cells that can be highlighted by immunohistochemical methods with antibodies to S-100 protein. Staining for cytoplasmic argyrophilia can be useful in diagnosis and often shows that the neoplastic chief cells contain myriad pinpoint cytoplasmic granules. This stain is seldom done today.
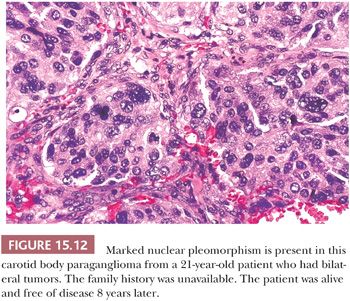
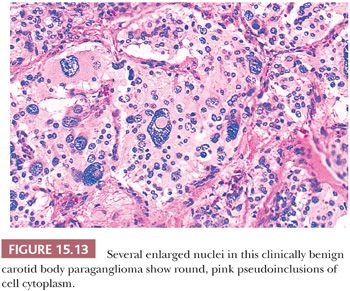
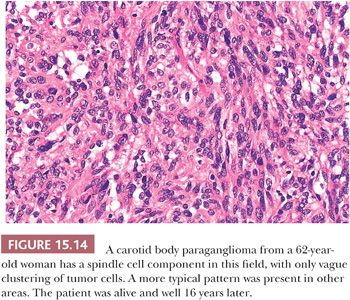
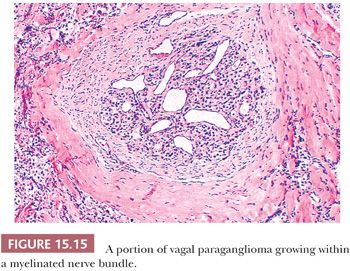
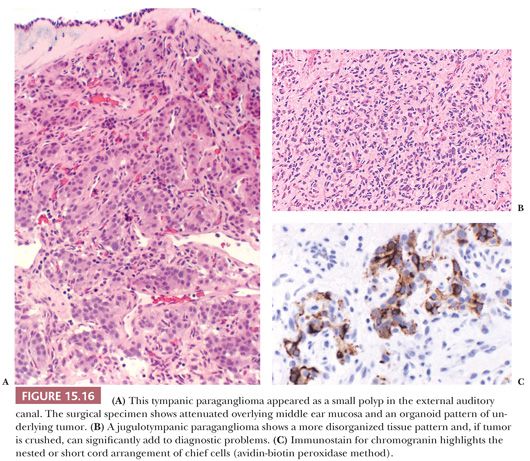
Nuclear hyperchromasia and pleomorphism can be quite prominent, but these are not reliable criteria for judging malignancy (Fig. 15.12) (34,40). Nuclear pseudoinclusions, representing invagination of cell cytoplasm, are occasionally seen (Fig. 15.13). A most unusual architectural pattern is a spindle cell or pseudosarcomatous arrangement of cells that, on careful examination, is usually accompanied by a more typical nesting pattern elsewhere in the tumor (Fig. 15.14) (34,91). Large expansile or locally aggressive tumors, such as CBPs or VPs, may incorporate myelinated nerve bundles, but true intraneural growth is seldom seen (Fig. 15.15). Limited biopsy material can hamper accurate diagnosis; an example is the small tympanic paraganglioma, which can appear as an aural polyp (Fig. 15.16A) or a small, hypercellular biopsy specimen of a skull base tumor without an obvious organoid or nesting pattern (Fig. 15.16B). Immunostain for chromogranin A may greatly aid in diagnosis by demonstrating discrete collections of neoplastic chief cells (Fig. 15.16C). It is well known that biopsies of paragangliomas can be hazardous and lead to brisk hemorrhage.
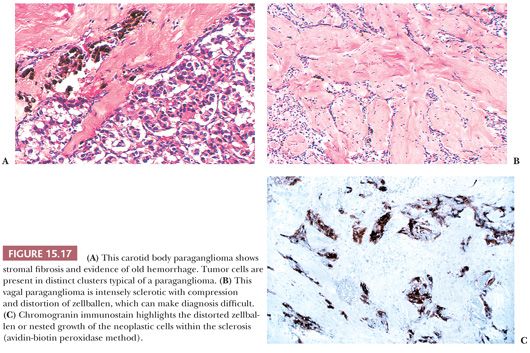
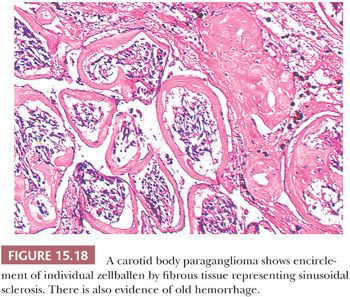
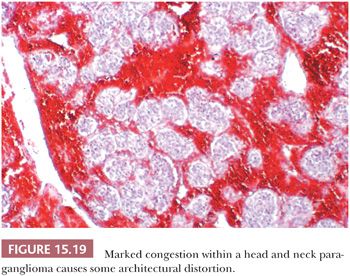
Stromal alterations may be apparent in some paragangliomas and, at times, are so marked as to obscure the true nature of the tumor (34,40). An example is stromal fibrosis with evidence of old hemorrhage (Fig. 15.17A) that may be so extensive that it causes compression and distortion of nests of neoplastic chief cells (Fig. 15.17B). In some cases, one may find sinusoidal sclerosis, which accentuates the organoid arrangement of tumor cells (Fig. 15.18). Congestion and hemorrhage within the tumor can cause wide spacing of individual zellballen (Fig. 15.19), but more typical diagnostic features can usually be found. Interstitial amyloid deposits were reported in two CBPs by Capella and Solcia (97) on ultrastructural study, but none was illustrated.
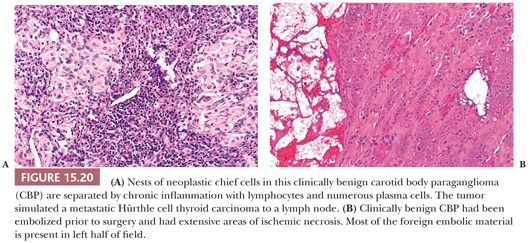
Dysmorphic vessels as a result of symmetric or asymmetric myointimal proliferation or thickening may be present, and in some tumors, vascular ectasia or an arborizing pattern can focally simulate a hemangiopericytoma. Chronic inflammatory infiltrates are uncommon. They may be evident as sparse perivascular lymphocytic infiltration, usually at the periphery of the tumor, but rarely, the inflammatory component can be more extensive, with separation of nests of chief cells. It can also simulate a lymph node metastasis, such as Hürthle cell carcinoma of the thyroid (Fig. 15.20A). Necrosis is an unusual morphologic finding, but it may be extensive and confluent if the tumor has been effectively embolized before surgery (Fig. 15.20B).
SYMPATHOADRENAL NEUROENDOCRINE SYSTEM
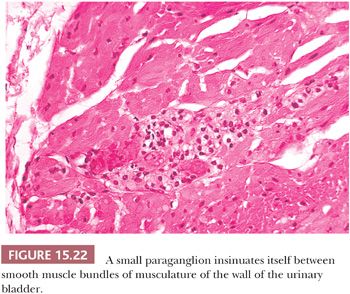
The prototype tissues are the adrenal medullae, which synthesize and secrete catecholamines, causing rapid physiologic changes that are dissipated rather quickly. The distribution of extra-adrenal paraganglia (Fig. 15.21) parallels that of the sympathetic nervous system within the abdomen and urinary bladder, thorax, and neck (98), and some paraganglia are located within viscera such as the gallbladder and urinary bladder (Fig. 15.22). During fetal life, the chromaffin cells of the adrenal gland are usually inconspicuous, but postnatally, they develop rapidly, becoming structurally similar to medullary cells of the adult gland by the end of the first year of life. Epinephrine is the predominant catecholamine in the normal adrenal medulla in a ratio of approximately 4:1 relative to norepinephrine (34).
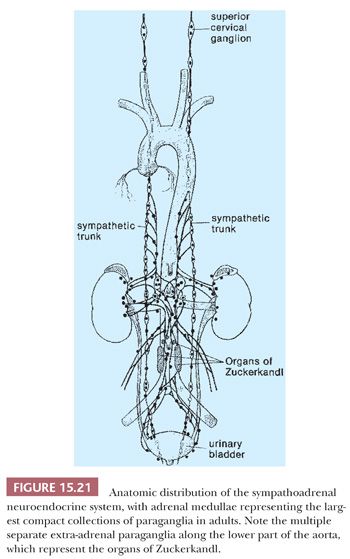
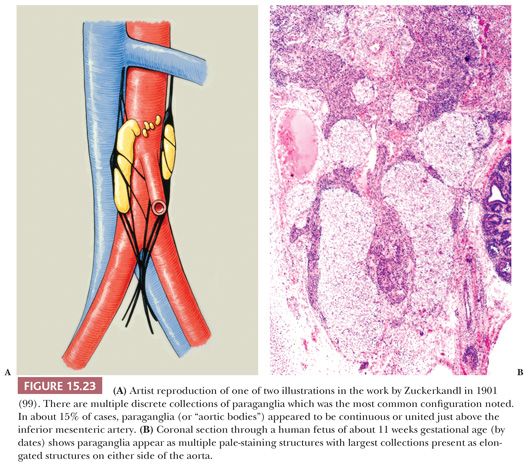
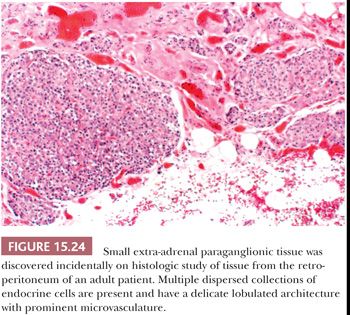
Most of the chromaffin tissue in the fetus is extra-adrenal in location, with the most prominent collections residing on either side of the aorta near the origin of the inferior mesenteric or renal arteries, down to the aortic bifurcation. These paraganglia were described by Zuckerkandl (99,100) in 1901 and referred to as the “aortic bodies” (Fig. 15.23A,B); on macroscopic examination, they may be difficult to distinguish from small lymph nodes or sympathetic ganglia (101). The organs of Zuckerkandl involute after birth; by the age of 6 or 7 years, only microscopic collections of chromaffin cells can be detected (Fig. 15.24) (102). The function of fetal chromaffin tissue in utero is not fully known, but it may have some role in maintenance of vascular tone and blood pressure. Norepinephrine predominates in extra-adrenal chromaffin tissue, such as the organs of Zuckerkandl, and catecholamine content is reported to decline with structural involution (103). The microanatomy of sympathetic paraganglia elsewhere in the abdomen, chest, and other locations is less well defined.
EXTRA-ADRENAL PARAGANGLIOMAS
These tumors arise predominantly in the retroperitoneum anywhere from the upper abdomen to the pelvic floor. One of the more common sites is the anatomic region corresponding to the organs of Zuckerkandl. It has been estimated that 5% to 10% of pheochromocytomas are extra-adrenal in location, mainly in the retroperitoneum but also in the posterior thorax and neck (34,40,104). A higher incidence of extra-adrenal tumors has been reported in childhood (20% to 25%) (105), with the most common locations being the retroperitoneum and head and neck region (106). In the literature review of extra-adrenal paragangliomas (mainly sympathoadrenal neuroendocrine system) by Fries and Chamberlin (107), 71% of tumors were located in the superior or inferior para-aortic area, 9.8% arose in the urinary bladder, 12% were intrathoracic, and 1.2% were cervical.
Other anatomic sites of origin include the urinary bladder (108), gallbladder (109), spermatic cord (110), prostate gland (111), prostatic urethra (112), pancreas (113), uterus (114), and renal hilus (115). A pigmented paraganglioma of the kidney has recently been reported (116). These extra-adrenal tumors are often hormonally active, with excess catecholamine secretion. It is worth noting that although functionally active CBPs and other head and neck paragangliomas rarely occur, some paragangliomas reported to be CBPs probably arise from the cervical sympathetic trunk (117,118); because of overlap in histologic features, the distinction may rest with precise anatomic localization. Multifocal paragangliomas can develop at various sites where one might expect to find paraganglionic tissue. An extreme example of paragangliomatosis was reported by Karasov et al. (119) in a patient who had 21 paragangliomas removed between 13 and 17 years of age and had evidence of additional tumors.
GROSS MORPHOLOGY
Most tumors are well circumscribed and, at times, almost encapsulated, ranging in diameter from only a few centimeters to 20.0 cm. The average diameter of extra-adrenal retroperitoneal tumors reported from Memorial Hospital was 9.9 cm, and the functionally active tumors tended to be smaller than the nonfunctional tumors (120). The average size of paragangliomas of the urinary bladder is approximately 2 cm (range, 0.3 to 5.5 cm) (121), and the average size of paravertebral paragangliomas is 5.8 cm (122). Some tumors are red-brown and show areas of hemorrhage on cross section, whereas other foci appear tan to gray-white with a slightly bulging surface. Hemorrhage and cystic degeneration may be marked in some tumors (Fig. 15.25).
MICROSCOPIC PATHOLOGY
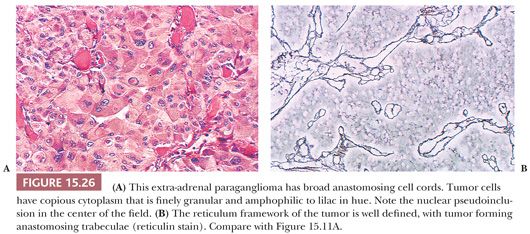
The most common architectural pattern in pheochromocytomas and extra-adrenal paragangliomas is an anastomosing cell cord or trabecular arrangement of tumor cells (Fig. 15.26A,B). In a minority of cases, one may see a more discrete, organoid pattern of oval to round clusters of cells similar to head and neck paragangliomas. Occasionally, there may be a solid or diffuse growth pattern or even a spindle cell component. Tumor cells have relatively abundant cytoplasm that is lightly acidophilic and finely granular, and some neoplasms may appear “oncocytic.” Occasionally, the cytoplasm has an amphophilic or lilac coloration. There may be considerable variation in nuclear size and shape with marked pleomorphism, but again, this is not a reliable feature in terms of diagnosing malignancy (34,40).
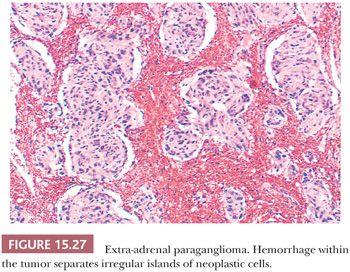
Nuclear pseudoinclusions are more frequently found in adrenal and extra-adrenal paragangliomas compared with head and neck paragangliomas (Fig. 15.26A); they have been shown to be invaginations of cell cytoplasm (123). Hemorrhage within the tumor can separate clusters of tumor cells (Fig. 15.27) or give a pseudopapillary or pseudoglandular pattern. The chromaffin reaction is positive in most of these tumors, provided the tumor is fixed immediately in the fresh state in appropriate fixatives, such as Orth, Zenker, or Helly fluid. In cases studied at the National Cancer Institute (NCI), the chromaffin reaction was positive for all sympathoadrenal paragangliomas when fixed appropriately. The reaction depends on the oxidation of epinephrine and norepinephrine to adrenochrome pigments. Today, the chromaffin reaction is largely of historic interest because it is not essential for confirming the diagnosis, having been replaced by more modern methods such as immunohistochemical stains.
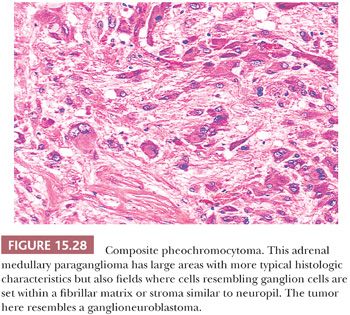
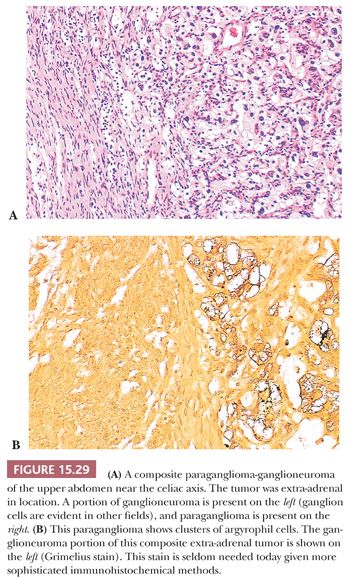
Ganglion-like cells may be present in some tumors. When accompanied by a fibrillary stroma or matrix resembling neuropil, the tumor may focally resemble a neuroblastoma or ganglioneuroblastoma and is referred to as a composite pheochromocytoma (Fig. 15.28) (34,40). Composite pheochromocytomas and composite extra-adrenal paragangliomas may also combine the histomorphologic features of a paraganglioma-ganglioneuroma (Fig. 15.29A,B) (124,125). A composite pheochromocytoma-ganglioneuroma has been reported in association with multiple endocrine neoplasia (MEN) type 2A (126). Composite pheochromocytoma–malignant peripheral nerve sheath tumor has also been reported (127). The occurrence of composite pheochromocytomas reflects the plastic phenotype of cells grown in vitro as well as the close relationship between the endocrine and nervous systems. Common embryogenesis from neural crest may help explain these unusual tumors (34). Rare cases of corticomedullary mixed tumors of the adrenal gland have also been reported (128).
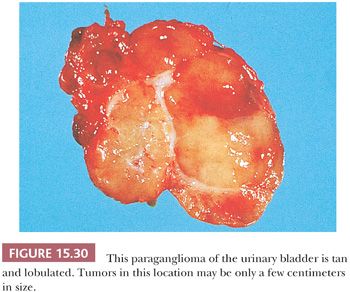
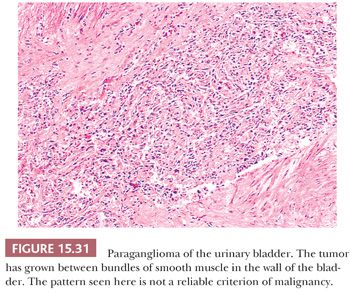
The urinary bladder can also be a primary site for a paraganglioma, and the patient may experience syncope or hypertension with bladder distention and/or micturition (34,121). These tumors are usually located in the bladder trigone or near the ureteral orifice, followed by the dome and lateral walls. The gross appearance is similar to paragangliomas elsewhere, except that the tumor tends to be smaller in size (Fig. 15.30). These neoplasms may show broad trabeculae growing between and separating bundles of smooth muscle, but this should not by itself be regarded as evidence of malignancy (Fig. 15.31). Occasional cases of malignant paragangliomas of the bladder have been reported (121,129). One must be aware that some invasive urothelial carcinomas may exhibit an organoid, trabecular, or nested growth pattern, and in these cases, the possibility of paraganglioma must enter in the differential diagnosis.
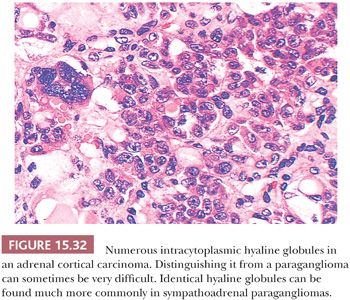
Intracytoplasmic hyaline globules are not uncommon; of more than 120 sympathoadrenal paragangliomas studied at the NCI, hyaline globules were identified in 47% of tumors (130). They were also noted in 43% of pheochromocytomas reported elsewhere (131). In some tumors, these hyaline globules are numerous, whereas in other neoplasms, they are very difficult to find on casual inspection. These globules have been related in some fashion to secretory activity by the tumor. It should be noted that virtually identical hyaline globules can be found in some adrenal cortical neoplasms, both benign and malignant—although much less frequently—and this may be a source of misdiagnosis of these tumors, which, in some instances, can be difficult to distinguish by routine histology from pheochromocytomas (Fig. 15.32) (132,133). Hyaline globules are extremely uncommon in head and neck paragangliomas such as CBPs.
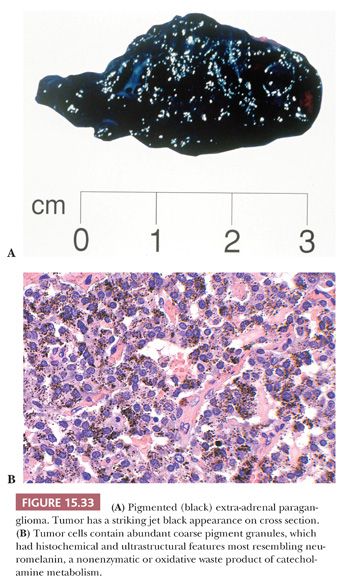
Interstitial amyloid has also been reported in a retroperitoneal paraganglioma (134) as well as pheochromocytomas (135). In one study, amyloid was identified in only 9% of pheochromocytomas (136). Pigmented or black pheochromocytomas and extra-adrenal paragangliomas have also been reported (34,137). The gross appearance can be striking, particularly when it has a jet-black color. Some tumors have not been rigorously investigated to distinguish the precise nature of the pigment, whereas in others, the presence of melanosomes or premelanosomes has been confirmed by ultrastructural examination. In one case, the intracytoplasmic pigment was abundant (Fig. 15.33) and had characteristics of neuromelanin (138). A recently reported case of pigmented renal paraganglioma also had pigment granules consistent with neuromelanin (116).
MULTICENTRIC AND FAMILIAL OCCURRENCE OF TUMORS
Since the report by Chase (139) of the familial occurrence of CBP, there have been numerous patients described with familial or hereditary paragangliomas in the head and neck region and paragangliomas of the sympathoadrenal neuroendocrine system (140). The complexities of the genetic background have grown considerably. Pheochromocytoma (PCC) and paraganglioma (PGL) are neuroendocrine tumors arising from adrenal medulla or extra-adrenal paraganglia respectively. Up to one-third of patients with PCC/PGL carry one of the germline mutations predisposing to the development of these tumors (141,142). Ten susceptibility genes have been identified for PCCs and PGLs (142). The genes NF1 (17q11.2), RET (10q11.2), VHL (3p25-26), and MAX (11q23) are associated with PCC (adrenal medullary PGL), and in cases of RET and MAX genes, the adrenal tumors may be bilateral. The predominant mode of inheritance is autosomal dominant, with two syndromes (PGL 1 and 2) having paternal inheritance (142). These genetic susceptibilities associated with PCCs will not be discussed further in this chapter. Familial PGL syndromes have been associated with germline mutations in genes encoding subunits of the SDH enzyme complex. SDH consists of the subunits SDHA, SDHB, SDHC, and SDHD which forms mitochondrial complex II and plays an important role in the mitochondrial respiratory chain and the tricarboxylic acid cycle. There are four recognized PGL syndromes: PGL 1 (SDHD, locus 11q23.1), PGL 2 (SDHAF2 [SHD5], locus 11q12.2), PGL 3 (SDHC, locus 1q23.3), and PGL 4 (SDHB, locus 1p36.1). PGL 5 (SDHA, locus 5q15) has a very low association with PCC/PGL, but has been linked with the Leigh syndrome, a familial neurologic disorder (141,142). Another susceptibility gene is TMEM127 (locus 2q11.2) (141,142).
PGL 1, 2, and 3 have been associated with PGLs of the head and neck region, and a number of these are multifocal. Currently, the role of genetic screening for SDH mutations is somewhat controversial. PGL 1 is the most common PGL syndrome and is associated with a low incidence of malignancy (<5%), whereas PGL 4 has the greatest association with malignant PCCs and malignant head and neck PGLs (142). In a recent study of patients with PCCs and PGLs, a retrospective review of 139 consecutive patients from a medical genetics clinic found a 41% overall mutation rate, indicating that these tumors were no longer the “tumor of tens” (i.e., 10% hereditary), and because the most common mutated gene was SDHB (highest risk of malignancy), the authors suggested that all patients with PCC/PGL undergo genetic testing (143). SDHB has also been associated with the development of renal neoplasms but at a lower penetrance. Recently, a new syndrome of PGL and somatostatinoma associated with polycythemia has been reported (144,145), and most recently, a case has been reported with bilateral PCC (146). It has been associated with somatic mutation in the HIF (hypoxia inducible factors) 2A gene.
ASSOCIATION WITH OTHER ENDOCRINE DISORDERS
CBPs have been reported in association with the triad described by Carney et al. (147) that consists of extra-adrenal PGL, gastric epithelioid leiomyosarcoma, and pulmonary chondroma. Carney triad (CT) primarily affects young females; in recent literature, the gastric epithelioid leiomyosarcoma component is now included in the gastrointestinal stromal tumor (GIST) category (148–150). Although initially the condition was thought to possibly be familial (151), a familial inheritance pattern has not been definitely confirmed. Recent molecular genetic studies have attempted to elucidate the pathogenetic pathway. In a review of 24 cases of CT, four patients had a CBP and three had additional tumors, including an APP that may have been functionally active (152). Head and neck PGLs have also been reported in association with papillary carcinoma of the thyroid (153) and hyperparathyroidism caused by either adenoma or hyperplasia (154,155). An unusual complex of tumors, including bilateral CBPs, raises the possibility of a new pattern of MEN syndrome (156,157). Additionally, a new syndrome of familial PGL and GIST, distinct from CT, has been identified; this has been referred to as the dyad of PGLs with GISTs or Carney-Stratakis syndrome (149).
CYTOLOGY AND FINE-NEEDLE ASPIRATION BIOPSY OF PARAGANGLIOMAS
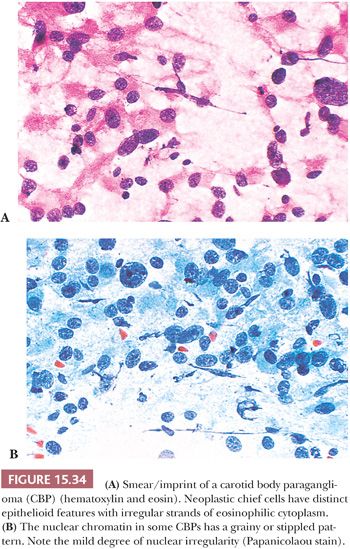
Smears or imprints of resected PGLs show irregular clusters of tumor cells with eosinophilic, finely granular cytoplasm and irregularly shaped nuclei that can have finely stippled or densely clumped chromatin (Fig. 15.34A,B). However, the correct diagnosis can be very difficult to make on cytologic evaluation alone, and the nuclear pleomorphism can be so marked that the unwary may be lured into making a diagnosis of malignancy. The cell cytoplasm of adjacent cells may be pulled apart into tapering irregular processes that give a latticelike configuration. Correlation with histologic findings will usually ensure an accurate diagnosis.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


