Papillomaviruses
Peter M. Howley
John T. Schiller
Douglas R. Lowy
The papillomaviruses (PVs) comprise a group of nonenveloped epitheliotropic DNA viruses that induce benign lesions of the skin (warts) and mucous membranes (condylomas). Some PVs have also been implicated in the development of epithelial malignancies, especially cancer of the uterine cervix, other tumors of the urogenital tract, and upper airway cancers. The recognition that PVs are an important cause of human cancer has led to the development of a preventive virus-like particle (VLP)–based vaccine targeted to the human papillomavirus (HPV) types most often found in the cancers. This chapter focuses primarily on the HPVs.
History
Warts were known to the ancient Greeks and Romans. Their infectious nature was recognized, but until the nineteenth century, genital warts were usually considered to be a form of syphilis or gonorrhea. The viral nature of human warts was demonstrated in the early 1900s when cell-free filtrates from lesions were shown to transmit the disease.110 PVs were subsequently identified in a variety of vertebrate species in addition to humans.346,592 Because PVs are species-specific in their host range, it has not been possible to study the biology of HPVs in animals.
The first animal PV was identified in the 1930s by Richard Shope, who characterized the transmissible nature of cutaneous papillomas arising in wild cottontail rabbits.553 The Shope papillomavirus, now designated the cottontail rabbit papillomavirus (CRPV), was the first DNA tumor virus identified. Shope’s research also showed that although systemic injection with papilloma suspensions did not produce detectable infection, it could induce serum neutralizing antibodies and protect rabbits against high-dose cutaneous viral challenge.552 These findings laid the groundwork for believing that a preventive vaccine against a PV could be based on the induction of humoral immunity. In addition to causing benign papillomas, some warts induced by CRPV were observed to undergo malignant progression,513,599 and for the next two decades, CRPV was an important model for the fundamental study of viral tumorigenesis.328,598 However, its use as a model tumor virus was largely supplanted by the discovery in the late 1950s of the polyomaviruses, which could replicate in cultured cells and induce morphologic transformation in vitro, in contrast to CRPV, and were tumorigenic for experimental animals.417
In addition to CRPV, the rabbit oral papillomavirus (ROPV) was also identified in the 1930s as a distinct virus of domestic rabbits.459 ROPV infected the oral mucosa of rabbits but not their skin, whereas CRPV had the opposite host range, and neither virus was infectious for heterologous hosts.
Furthermore, ROPV was not oncogenic, in contrast to CRPV, and neutralizing antibodies against one virus were shown not to be neutralizing for the other. The research on CRPV and ROPV thus established the multiplicity of PVs, the narrow host range of PVs to sites with stratified squamous epithelia that were cornified (skin) or noncornified (mucosa), and the notion that protection against one PV may not confer protection against another PV.
Furthermore, ROPV was not oncogenic, in contrast to CRPV, and neutralizing antibodies against one virus were shown not to be neutralizing for the other. The research on CRPV and ROPV thus established the multiplicity of PVs, the narrow host range of PVs to sites with stratified squamous epithelia that were cornified (skin) or noncornified (mucosa), and the notion that protection against one PV may not confer protection against another PV.
Although the PVs were studied less intensively in the 1950s and 1960s, that period was associated with some important advances, including the physicochemical analysis of PV virions and the demonstration that PV replication was associated with the differentiation process of the infected epithelium.379,515 However, it was the advent of molecular cloning in the 1970s that initiated more extensive studies of PVs. This technical advance enabled investigators to partially circumvent the inability to culture PVs, as the cloning of PV genomes greatly enhanced the ability to study their biologic and biochemical properties. The sequencing of the cloned PV genomes led to the identification of open reading frames (ORFs) as putative viral genes and permitted investigators to determine the function of viral genes by reverse genetics, resulting in a much wider interest in PV research.97,125,126 The bovine papillomavirus type 1 (BPV1) represented the standard PV for initiating these studies because the virus induced focal transformation of established rodent cell lines.45,163 The molecular cloning of the HPV genomes also led to the recognition that there were multiple HPV genotypes, and that a subset of these types was closely associated with human cancers, including cervical cancer.56,162,446 The appreciation of their medical importance, combined with improved tools for analyzing PVs, further enhanced the utility of PVs as a model of viral tumorigenesis. Although the study of animal PVs continues to bring new information to the field, the medical importance of HPVs has shifted emphasis toward the analysis of HPV, especially when it was established that the biochemical properties of some nonstructural viral proteins differed from those of their BPV-1 counterparts.165,525
Classification
Historically, the PVs were classified together with the polyomaviruses as a single family, the Papovaviridae. This grouping arose because although PV genomes and virions are larger than those of polyomaviruses, the viruses share many features, including a double-stranded circular DNA genome, an icosahedral capsid composed of 72 pentamers, a nonenveloped virion, and the nucleus as the site of viral replication and virion assembly. However, sequencing of PV genomes indicated that although PVs share a common genetic organization, they differ from that of polyomaviruses and have no major sequence homology to polyomaviruses, and PV transcription is unidirectional, in contrast to the bidirectional transcription of polyomaviruses. Recognition of these differences, and others, have led to PVs being designated as a distinct family, the Papillomaviridae, by the International Committee on the Taxonomy of Viruses (ICTV) in 2000.628
PVs have been isolated from many mammalian host species, birds, and reptiles, but thus far have not been identified in nonvertebrates. There are hundreds of PV types (as defined in the next paragraph). PVs are species-specific and many different PV types can infect a given host species. HPVs have been analyzed most intensively; by 2010, 120 different HPV types had been identified, and there are likely to be more.39
PVs are classified primarily according to the host species they infect and have been traditionally referred to as types based on their DNA sequence. A distinct type is one whose L1 DNA sequence is at least 10% different from that of other HPV types. The PV genomes have been organized phylogenetically based on their DNA sequence (Fig. 54.1; 44), according to the comparative homology of the L1 ORF. Similar phylogenetic relationships are also seen when homologies between other regions of the genome are compared, as PVs appear to have arisen primarily via point mutations scattered throughout the genome, rather than via recombination between PVs.91 These similarities are consistent with the conclusion that PVs have accompanied their host species during evolution and have evolved with them.40 Although all PVs share a similar genetic organization, the L1 DNA sequence identity is just over 40% between the most divergent genomes. On the other hand, two very closely related isolates may differ by only a single nucleotide. The current classification attempts to provide logical designations to cover this wide range of homology: genus, species, type, subtype, and variant. The broadest category is a genus. PVs are divided into 29 genera, each of which is designated by a letter of the Greek alphabet (Fig. 54.1). Within a given genus, the L1 DNAs of all members share more than 60% identity; conversely, they have less than 60% identity with members of other genera. A species is designated for those PVs within a given genus that share 60% to 70% identity. A viral type within a species has 71% to 89% identity with other types within the species. Within a type, there can be subtypes, which share 90% to 98% identity, and variants, which have more than 98% identity. Although there are relatively few subtypes,40 many variants have been identified for HPV-16, the type that has been examined in greatest detail because of its medical importance.100
Using this classification, HPVs are clustered among 5 of the genera: alpha, beta, gamma, mu, and nu, with the other 24 genera being occupied exclusively by animal PVs.39 The host species associated with each PV genus tend to be closely related evolutionarily. Therefore, PVs that infect nonhuman primates are found within the genera that include HPVs, and some HPVs are more closely related to nonhuman primate viruses than to some of the other HPVs in the genus. The HPVs of greatest medical importance, that is, those that are associated with genital and mucosal cancers, are members of the alpha genus. Most alpha PVs primarily infect genital and nongenital mucosal surfaces and the external genitalia. This group of PVs is often referred to collectively as the “genital-mucosal” types. The types that are associated with cervical cancer, often designated as “high-risk” types, are found in species 5,6,7,9, and 11.423,529 HPV16, the type found most frequently in cervical cancer, is a member of species 9, whereas the next most common cancer-associated type, HPV18, is a member of species 7. HPV-6, which causes most cutaneous genital warts, is a species 10 member.
In contrast to most species of the alpha genus, members of alpha species 4 (HPV2, −HPV27, and HPV57) are primarily infectious for nongenital skin. The beta, gamma, mu, and nu viruses also infect nongenital skin. The beta HPVs
include those that are often designated epidermodysplasia verruciformis (EV) specific, because they cause lesions mainly in patients with EV, a genetic susceptibility to widespread nongenital HPV lesions. Some PVs, including many members of the beta and gamma species, may behave as commensal agents, as they are frequently isolated from normal skin or plucked hair from humans and animals.12,59
include those that are often designated epidermodysplasia verruciformis (EV) specific, because they cause lesions mainly in patients with EV, a genetic susceptibility to widespread nongenital HPV lesions. Some PVs, including many members of the beta and gamma species, may behave as commensal agents, as they are frequently isolated from normal skin or plucked hair from humans and animals.12,59
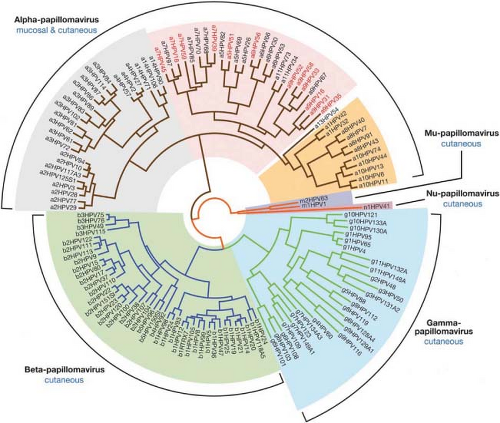 Figure 54.1. Phylogenetic tree demonstrating the evolutionary relationship among human papillomaviruses (HPVs). HPVs comprise five groups with different epithelial tropisms and disease associations. The alpha papillomaviruses include the low-risk mucosal types (many of which are within the orange-shaded branch) that cause genital warts, and the high-risk mucosal types (contained within the branch highlighted with pink shading) associated with anogenital preneoplasias and cancers. Although the cutaneous HPV types—most of which are contained within the gray (alpha), green (beta), and blue (gamma) shaded branches—are not generally associated with cancers, certain beta types have been implicated in the development of nonmelanoma skin cancers (NMSC) in immunosuppressed individuals and in epidermodysplasia verruciformis (EV) patients. The lower case letter and number preceding the HPV type refer to its genus and species. (Generated by John Doorbar; reprinted from Doorbar J, Quint W, Banks L, et al. The biology and life-cycle of human papillomaviruses. Vaccine 2012;30(Suppl 5):F55–F70, with permission.) |
The PVs in the delta genus, which include BPV1 and some other PVs of ungulates, cause fibropapillomas, rather than papillomas. This distinct pathology results from a proliferative dermal fibroblastic component under the epithelial portion of the lesion, because members of this genus induce nonproductive transformation of the fibroblasts, in addition to the productive infection of the overlying epithelium. The ability to transform nonepithelial cells is not species-specific. It can lead to the induction of nonproductive fibroblastic tumors in heterologous hosts under natural conditions, as in equine sarcoid of horses (from BPV1 or BPV2), or experimental hosts, such as hamsters. It also endows viruses such as BPV1 and BPV2 with the ability to induce focal transformation of cultured rodent cells.
Virion Structure
Papillomaviruses are small, nonenveloped, icosahedral DNA viruses that replicate in the nucleus of squamous epithelial cells. The PV particles are approximately 60 nm in diameter (Fig. 54.2). The virion particles consist of a single molecule of double-stranded circular DNA about 8,000 base pairs (bp) in size, contained within a spherical protein coat, or capsid, and composed two viral proteins L1 and L2. The DNA constitutes approximately 12% of the virion by weight, accounting for their density in cesium chloride of 1.34 g/mL.116
Fine structural analysis by cryoelectron microscopy (cryo-EM) and three-dimensional image reconstruction techniques has revealed that the viruses consist of 72 pentameric capsomers arranged on a T = 7 surface lattice.22,621 The capsomers comprise five L1 molecules with an L2 molecule occupying the axial lumen.73 As with the polyomavirus capsids, the capsomers exist in two environments, one capable of making contact with six neighbors as observed in the 60 hexavalent capsomers and the other with five neighbors in the 12 pentavalent vertex capsomers (Fig. 54.3). Analysis of proteins in the virus particle
showed that the viral DNA is associated with cellular histones to form a chromatin-like complex.172,469
showed that the viral DNA is associated with cellular histones to form a chromatin-like complex.172,469
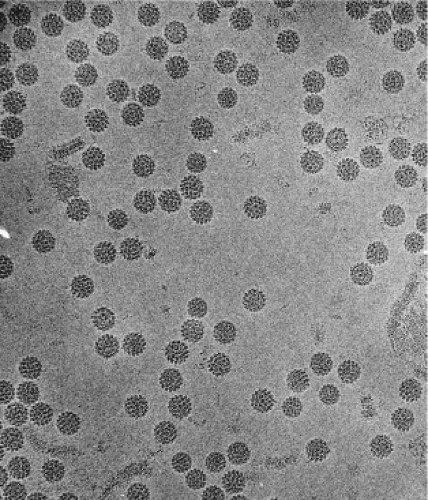 Figure 54.2. Electron micrograph of bovine papillomavirus 1 (BPV1) virion particles (55 nm in diameter). (Reprinted from Baker TS, Newcomb WW, Olson NH, et al. Structures of bovine and human papillomaviruses—analysis by cryoelectron microscopy and three-dimensional image reconstruction. Biophys J 1991;60:1445–1456, with permission.) |
VLPs can be produced from different PVs by expressing L1 alone using mammalian or nonmammalian expression systems.234,311,511 The morphology of VLPs containing only L1 appears identical to intact virus particles in low-resolution cryo-EM reconstructions.233 The structure of a truncated T = 1 HPV16 L1 VLP containing 12 pentamers has been solved by x-ray crystallography to 3.5 Å resolution.99 The structure of full-size BPV1 virions was recently solved to 3.6 Å resolution using cryo-EM.668
Genome Structure and Organization
The genomes of many of the human and animal papillomaviruses have been sequenced in their entirety, and the genomic organization of each of the papillomaviruses is similar. One characteristic of the genomic organization of all papillomaviruses is that all of the ORFs are located on one strand of the viral DNA, thus indicating that all of the viral genes are located on one strand. Transcriptional studies indicate that only one strand serves as a template for transcription.
The coding strand contains approximately 10 designated translational ORFs that are classified as either early (E) or late (L) ORFs, based on their location in the genome. The early region of the papillomavirus genomes encodes viral regulatory proteins including those viral proteins that are necessary for initiating viral DNA replication. The L1 and L2 ORFs encode the viral capsid proteins and are expressed only in productively infected cells.21 The position, size, and function of many of the ORFs are well conserved among the various PVs that have been sequenced and studied in detail thus far. The functions of the individual ORFs, the functions of which have been well characterized, are described in more detail in the appropriate sections of this chapter.
There is a region, of approximately one kilobase, in each of the papillomavirus genomes that contains no ORFs. This region has been referred to by several terms including the long control region (LCR), the upstream regulatory region (the URR), and the noncoding region. This region contains the origin of DNA replication as well as important transcription control elements. The genomic organization of HPV16 is shown in Figure 54.4.
Virus Replication
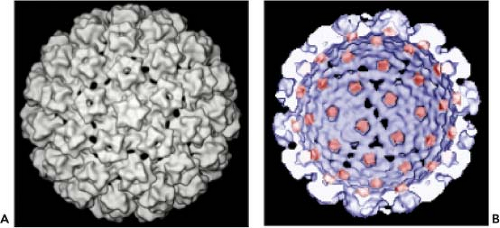 Figure 54.3. A: Three-dimensional (3D) reconstruction of a BPV virion viewed down a fivefold axis.621 B: 3D reconstruction of an interior/cutaway view of an human papillomavirus (HPV) L1/L2 virus-like particle (VLP) with the L2 specific density shown in red. (Adapted from Buck CB, Cheng N, Thompson CD, et al. Arrangement of L2 within the papillomavirus capsid. J Virol 2008;82:5190–5197.) |
The PVs are highly species-specific and have a specific tropism for squamous epithelial cells. The productive infection of cells
by the PVs can be divided into early and late stages. These stages are linked to the differentiation state of the epithelial cell. The specific tropism of the PVs for squamous epithelial cells is evidenced by the restriction of the viral replication functions, such as vegetative viral DNA synthesis, the production of viral capsid proteins, and the assembly of virions to differentiated epithelial cells. The close link of the papillomavirus life cycle with the differentiation program of the squamous epithelium is depicted in Figure 54.5.
by the PVs can be divided into early and late stages. These stages are linked to the differentiation state of the epithelial cell. The specific tropism of the PVs for squamous epithelial cells is evidenced by the restriction of the viral replication functions, such as vegetative viral DNA synthesis, the production of viral capsid proteins, and the assembly of virions to differentiated epithelial cells. The close link of the papillomavirus life cycle with the differentiation program of the squamous epithelium is depicted in Figure 54.5.
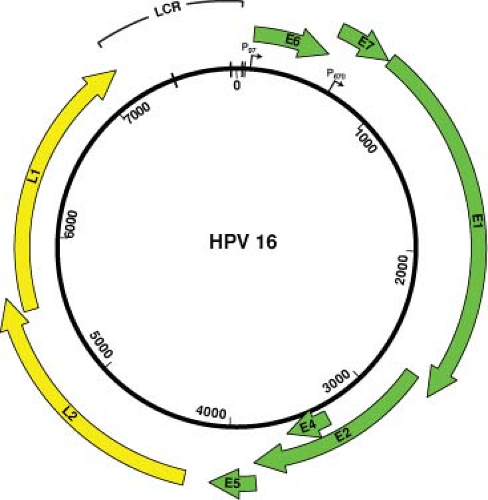 Figure 54.4. HPV16 genomic map. The numbers inside the circle indicate the nucleotide positions. The individual open-reading frames (ORFs) of the early (E) and late (L) regions are depicted as areas outside the double-stranded circular genome. Only one strand is transcribed, and transcription occurs in the clockwise direction. The early promoter (P97) is indicated by an arrow at the approximate nucleotide position of the RNA initiation site. P670 is the late promoter whose initiation sites map within the E7 ORF. The long control region (LCR) designates the long control region that contains the origin of DNA replication. The hatch marks indicate the four E2 binding sites within the LCR. |
The basal cell is the only cell in the squamous epithelium capable of undergoing cell division. Therefore, the virus must infect the basal cell in order to establish a persistent lesion. By in situ hybridization, it has been demonstrated that the viral DNA is indeed present within the basal cells and the parabasal cells of a papilloma.536 Furthermore, using probes to the early gene regions of the PVs, viral transcripts have been detected in the basal cells of the epidermis,584 and at least some early viral protein is found in basal cells.79 Late gene expression, synthesis of capsid proteins, vegetative viral DNA synthesis, and assembly of virions occur only in terminally differentiating squamous epithelial cells.
Virion Attachment, Entry, and Trafficking
Papillomaviruses have a unique infectious process that is intimately linked with their life cycle in stratified squamous epithelia. As noted above, productive papillomavirus infection is thought to require infection of the basal layer cells of the epithelium.152 To achieve selective infection of basal keratinocytes, the virions have evolved to preferentially bind initially to heparan sulfate proteoglycans (HSPGs) on the basement membrane exposed at sites of epithelial trauma or permeabilization, rather than to cells (Fig. 54.6).285,500 In vitro, the capsids bind directly to most epithelial cell lines in an HSPG-dependent manner.212 This difference probably reflects the adaption of in vitro propagated epithelial cells to the expression of HSPGs with modifications that resemble those that are normally found only on basement membrane in vivo.130 Basement membrane binding induces a conformational change in the capsid that exposes a highly conserved N-terminal L2 peptide motif to cleavage by furin or the closely related proprotein convertase 5/6.310 According to one model, cleavage induces a conformational change that exposes a capsid-binding site (probably on an L1 surface) for an as yet unidentified cell-surface receptor on keratinocytes and other cell types.310 Alternatively, a recently proposed model suggests that the virions may interact with cell surfaces as high molecular weight complexes also containing cleaved HSPGs and bound growth factors through an interaction with a variety of growth factor receptors.594 There is a remarkably long delay between cell surface binding and viral genome transcription of 1 to 3 days, both in vivo and in vitro.129,500 Internalization of the capsids from the cell surface takes at least 2 to 4 hours, and is very asynchronous, with some capsids remaining on the surface for a much longer time.123 In vitro studies indicate that the capsids are transported on the cell surface from filopodia at the leading edge of migrating cells to the central cell body via linkage to actin retrograde flow.526
The endocytic pathway involved in internalization and intracellular trafficking by PV capsids is controversial. Most studies have observed trafficking to acidified late endosomes either via clathrin-dependent or clathrin-independent uptake (Fig. 54.7).57,129,561 However, one study has implicated caveolin-dependent uptake with eventual trafficking to the endoplasmic reticulum,347 whereas another has suggested that tetraspanin-enriched microdomains may be involved in endocytosis.575 Several cell factors have been implicated in internalization and trafficking. These include cyclophilin B, a peptidyl-prolyl cis/trans isomerase, being required for exposure of the furin cleavage site in L242; sorting nexin 17, an adaptor protein involved in endosome cycling that inhibits routing of the capsids to lysosomes35; and FAK, a focal adhesion kinase.1 At least partial uncoating occurs in Lamp-2–positive late endosomes, but not until at least 8 to 12 hours after cell surface binding.127 L2–genome complexes escape the late endosomes, whereas the genomes packaged in L1-only capsids do not.293 A conserved C-terminal L2 peptide with strong membrane penetrating and disrupting activity in vitro may be directly involved in endosomolysis. In addition, the activity of the transmembrane protease γ-secretase is required for infection, perhaps functioning in endosome escape.296 Movement through the cytoplasm to the nucleus likely occurs along microtubules in association with the motor protein dynein.183 The fact that infection, at least in vitro, requires cell division has led to the conjecture that entry of the L2–genome complex into the nucleus may require nuclear envelope breakdown during mitosis, rather than active transport through nuclear pores.485 After cell division, the L2–genome complexes predominantly localize to specific nuclear structures, N10 bodies (also designated promyelocytic leukemia protein (PML) oncogenic domains, or
PODs).127 This localization promotes transcription of the viral genome. Potentiation of PV infection by association with ND10 bodies contrasts with the inhibitory activity of these structures in infections by herpes viruses, which target PMLs for degradation early in infection.171
PODs).127 This localization promotes transcription of the viral genome. Potentiation of PV infection by association with ND10 bodies contrasts with the inhibitory activity of these structures in infections by herpes viruses, which target PMLs for degradation early in infection.171
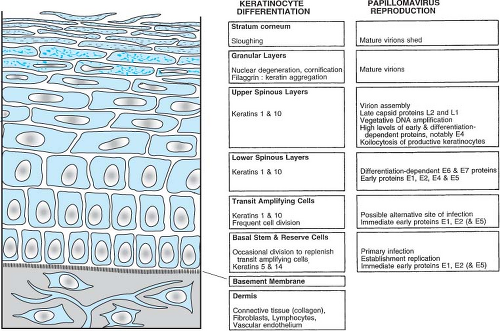 Figure 54.5. Differentiation of normal cutaneous squamous epithelium and papillomaviral activities in productively infected benign lesions. The various epithelial strata and the host differentiation, stage-specific, gene-expression profile are indicated in the left and center panels. In nonkeratinized squamous epithelia, such as cervical or laryngeal, keratins 4 and 13 are expressed in the place of keratins 1 and 10 in the differentiated cells. Although profilaggrin is expressed, there is no granular layer or stratum corneum in nonkeratinized squamous epithelia. The viral activities in the corresponding strata during productive infection shown on the right have been determined or inferred from in situ hybridization studies. (Reproduced from Chow LT, Broker TR. Small DNA tumor viruses. In Nathanson N, ed., Viral Pathogenesis. Philadelphia: Lippincott-Raven, 1997:267–301.) |
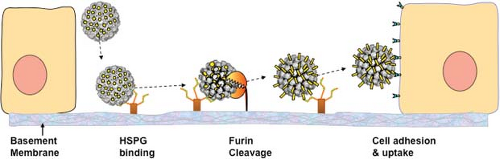 Figure 54.6. Model of in vivo papillomavirus infection. The virion first binds to heparan sulphate proteoglycans (HSPGs) on the basement membrane exposed after disruption. This induces a conformational change exposing a site on L2 (depicted in yellow) susceptible to proprotein convertase (furin or PC5/6) cleavage. After L2 cleavage, an L2 neutralizing epitope is exposed and a previously unexposed region of L1 binds to an unidentified secondary receptor on the invading edge of the epithelial cells. |
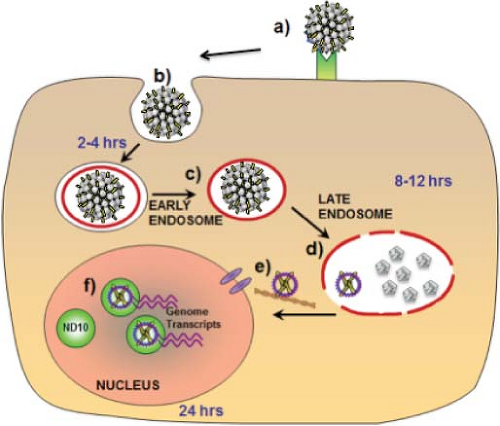 Figure 54.7. Infectious process after cell binding. After binding to a cell surface receptor (a), the virus enters the cell via an endocytic pathway (b) and within 4 hours localizes in the early endosome (c). By 12 hours the virus uncoats within the late endosome and the viral genome complexed with L2 is released (d). The L2–genome complex traffics through the cytoplasm, perhaps via microtubules, and enters the nucleus by 24 hours (e). After nuclear entry, the complex co-localizes with ND10 and viral genome transcription begins (f). |
Viral Transcription
The replicative phase of the papillomavirus life cycle is tightly linked to the differentiation program of the squamous epithelium. Historically, BPV1 served as the prototype for analyzing the papillomavirus transcription program. The studies have been carried out in a variety of systems, including viral RNAs from rodent cells transformed by BPV1 as well as those from infected wart tissues. More recently viral transcription studies have been extended to some of the HPVs associated with genital tract lesions—such as HPV11, HPV16, HPV18, and HPV31—by using HPV-positive clinical lesions, xenograft tissue in nude mice, cervical carcinoma cell lines, as well as organotypic culture systems. This portion of the chapter on transcription focuses largely on HPV16 and HPV31, both of which have been extensively analyzed by in vitro culture techniques.411
Viral RNAs and Promoters
Papillomavirus transcription is complex due to the presence of multiple promoters, to alternate and multiple splice patterns, and to the differential production of messenger RNA (mRNA) species in different cells.
A transcription map of HPV31 is shown in Figure 54.8. As with BPV1, multiple promoters are involved in generating the various mRNA species for the genital tract HPVs. For HPV31, P97 is the major promoter active in nonterminally differentiated cells. This promoter, which directs the expression of E6 and E7 as well as several other early gene products, is analogous to P97 of HPV16 and P105 of HPV18. Upon differentiation of immortalized keratinocytes harboring episomal HPV-31 DNA, there is activation of the differentiation-dependent, late promoters, P670 for HPV16 and P742 for HPV31, that direct the expression of the late gene products, including E4, L1, and L2, as well as an increase in the level of the E1 mRNA.321,517
An important difference in the structures of the E6 and E7 mRNAs and in the manner by which they are expressed distinguishes the “high risk” and “low risk” HPV types. For the “high risk” HPVs such as HPV16 and HPV18, a single promoter (P97 for HPV16 and HPV31, or P105 for HPV18) directs the synthesis of mRNAs with E6 and E7 intact or with splices in the E6 gene (Fig. 54.8). The species with E6 intact could be translated into E6 but not E7 since there is insufficient spacing for translation reinitiation. The mRNAs with the spliced E6 splice the 5′ end of the E6 ORF (referred to as E6*) to a translation frame with stop codons that provide sufficient spacing for translation reinitiation of the E7 ORF and are therefore likely to represent the E7 mRNAs. In contrast, the E6 and E7 genes of the “low risk” HPVs such as HPV6 and HPV11 are expressed from two independent promoters.105
Regulation of Transcription (Cis Elements)
Papillomavirus transcription is tightly regulated by the differentiation state of the infected squamous epithelial cell. This is evident from the analysis of the differential expression of viral RNAs in cells from the different levels of the epithelium in warts.107 It is also evident from studies of infected keratinocytes using organotypic and suspension tissue culture systems that permit epithelial cell differentiation.149,321,410 Proper transcription of the viral genome is dependent upon the differentiation state of the epithelial cells and on cellular transcription, splicing, and other RNA processing factors.373
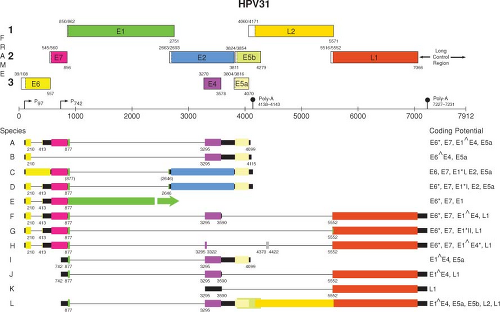 Figure 54.8. Transcription map of HPV31.266,267,321,448 A linearized version of the genomic map is shown at the top. Transcripts initiated at the early viral promoter, designated P97, are expressed in the nonterminally differentiated cells in the lower portion of the epithelium, whereas those initiating from the late promoter (P742) are expressed upon differentiation in the cells committed to the replication of the progeny virions. (Reproduced from the PAVE transcription maps, http://pave.niaid.nih.gov.easyaccess1.lib.cuhk.edu.hk) |
The LCR (also referred to as the URR) region of papillomavirus contains enhancer elements that are responsive to cellular factors as well as to virally encoded transcriptional regulatory factors. Each of the viral LCRs that have been studied in detail have been found to contain constitutive enhancer elements that have some tissue or cell-type specificity. These constitutive enhancer elements play an important role for the initial expression of the viral genes after virus infection and may also be important in the maintenance of viral latency. A number of transcription factor binding sites have been identified in the LCRs of the various papillomaviruses that have been carefully studied. Included among them are sites that bind AP1, SP1, Oct-1, and YY1, among others.38,373 The HPV16 LCR has also been shown to contain nuclear matrix attachment sites that may be important for controlling viral gene expression.604 In addition to the binding sites for cellular transcription factors, the LCR contains binding sites for the virally encoded E2 regulatory proteins and the origin of DNA replication that binds the E1 replication factor.
E2 Regulatory Proteins
The papillomavirus E2 proteins have well-characterized regulatory functions affecting viral transcription, viral DNA replication, and long-term plasmid maintenance. E2 was first described as a transcriptional activator572 capable of activating viral transcription through E2 responsive elements located within the viral genome.571 The E2 proteins are relatively well conserved among the papillomaviruses in two domains: a sequence-specific DNA-binding and dimerization domain located in the carboxy terminal region of the protein and a transactivating domain that is located within the amino terminal half of the protein.211,398 These two domains are separated by an internal hinge region that is not well conserved in size or in amino acid composition among different papillomaviruses. The E2 proteins bind the consensus sequence, ACCN6GGT,9,362 and can regulate transcription from promoters containing E2 binding sites.242,251,570 E2 binds ACCN6GGT motifs as a dimer; the DNA binding dimerization domain localizes to the carboxy terminus of E2.399
The E2 proteins have been best studied in the BPV system, where three species have been identified (Fig. 54.9). The full-length protein (E2TA) can function as a transactivator or a repressor depending on the location of the E2-binding sites within the enhancer/promoter region. The two shorter forms of E2 called E2TR and E8/E2 have been described as repressors because they can inhibit the transactivation function of the full-length E2TA.344,345 The shorter E2 proteins contain the DNA binding and dimerization domains of the C terminus but lack the transactivation domain. E2TR and E8/E2 can inhibit the transcriptional transactivating function of the full-length polypeptide by competing for its cognate DNA binding sites and by forming inactive heterodimers with the full-length transactivator protein. The crystal structure of the
dimeric DNA-binding domain of BPV1 E2 revealed a previously unobserved structure for a DNA-binding protein, which is a dimeric antiparallel β barrel.246 The crystal structure of the N-terminal transactivation domain has also been resolved for HPV16 and HPV18.14,240 The structural studies of the E2 N-terminal domain indicated that it could form a dimer both in the crystal and in solution. Because amino acids that are necessary for transactivation are located at the dimer interface, the dimer structure may be important in the interactions of E2 with viral and cellular transcription factors. The dimer formation may contribute to the stabilization of DNA loops, which may serve to relocate distal DNA-binding transcription factors to the site of human papillomavirus transcription initiation.14
dimeric DNA-binding domain of BPV1 E2 revealed a previously unobserved structure for a DNA-binding protein, which is a dimeric antiparallel β barrel.246 The crystal structure of the N-terminal transactivation domain has also been resolved for HPV16 and HPV18.14,240 The structural studies of the E2 N-terminal domain indicated that it could form a dimer both in the crystal and in solution. Because amino acids that are necessary for transactivation are located at the dimer interface, the dimer structure may be important in the interactions of E2 with viral and cellular transcription factors. The dimer formation may contribute to the stabilization of DNA loops, which may serve to relocate distal DNA-binding transcription factors to the site of human papillomavirus transcription initiation.14
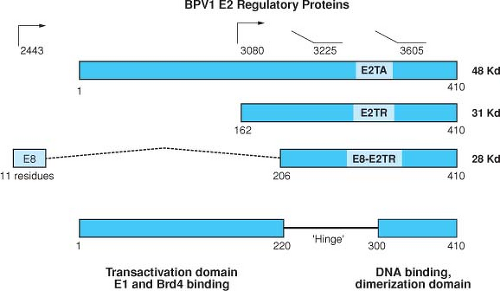 Figure 54.9. Structure of the BPV1 E2 gene products. The structures of the three known proteins encoded by the BPV1 E2 ORF are indicated. The 48-kD full-length E2 transactivator can be expressed from an unspliced message from P2443 or from a spliced messenger RNA (mRNA) from upstream promoters by utilizing a splice acceptor at nT 2558. The 31-kD and 28-kD forms of the repressor are expressed from P3080 and as an E8/E2 fusion by a spliced mRNA as shown, respectively. The transactivation domain consists of a region of approximately 200 amino acids at the N-terminal region of the full-length E2 protein that is relatively well conserved among papillomaviruses. This region, which is acidic and is predicted to contain amphipathic helices, is only present in the full length form of E2. The N-terminus of E2 contains the site for binding E1. The 110 C-terminal amino acids are also conserved and comprise the DNA-binding and dimerization domain. The basic region and the hydrophobic repeats are indicated.400 |
Mutations in the BPV1 E2 ORF have pleiotropic effects, disrupting transformation, replication, and transcriptional functions. Studies have shown that expression of the early region viral genes is under the control of the viral E2 gene product through E2 responsive elements located within the viral LCR. The actual role of the E2 transactivation function in the PV life cycle remains to be elucidated, however. Studies with an HPV31 genome that carried a mutant E2 gene that was defective for transactivation but competent for DNA replication competent could still be established as a stable episome and could induce differentiation-dependent late functions.589
E2 transcriptional regulation has also been well studied for the genital tract-associated HPVs. The binding of E2 to its cognate sites within the LCR of the HPV genomes results in the modulation of viral promoter activity. The E6 and E7 transforming genes of HPV16 and HPV18 are transcribed from the major early promoter (P97 and P105, respectively) contained within the LCR of their respective genomes. In human epithelial cells, the HPV16 P97 promoter and the HPV18 P105 promoter display basal activities that can be repressed by full-length E2.37,506,608,609 There are four E2-binding sites upstream of the P97 and P105 promoters that mediate this repression. In addition to binding at its cognate sites, the E2 transcriptional activation function is required for E2-mediated promoter repression. Specific conservative point mutations within the bovine or human E2 transactivation domain that eliminate E2-mediated transcriptional activation also eliminate E6/E7 promoter repression.219,436 The bromodomain containing protein 4 (Brd4) is involved in this transcriptional repression.677 An unbiased whole genome small interfering RNA (siRNA) screen validated the involvement of Brd4 in this transcriptional repression and also revealed independent roles for the histone demethylase known as SMCX as well as components of the TIP60 histone acetylase complex including EP400.560
E2 can suppress the growth of HPV-positive cervical cancer cell lines through the transcriptional repression of the viral E6 and E7 genes.156,188,269,436,609 This repression results in the reactivation of the Rb and p53 tumor suppressor pathways that are inhibited by E6 and E7 and induce a cell cycle arrest and cellular senescence.217,218,649
E2 is a multifunctional protein. Its functions as a transcriptional activator and repressor are likely mediated by interactions with specific cellular factors, some of which have now been identified (Table 54.1). In addition to its role as a transcriptional regulator, E2 has critical role in viral DNA replication. The full-length E2 proteins are critical auxiliary factors for viral DNA replication.103,137,623 This aspect has been
best studied in the BPV system, where E2 was first shown to complex with E1 and to strengthen the affinity of E1 for binding to the origin of DNA replication.416,623
best studied in the BPV system, where E2 was first shown to complex with E1 and to strengthen the affinity of E1 for binding to the origin of DNA replication.416,623
Table 54.1 Cellular Targets and Functions of the Papillomavirus E2 Regulatory Proteins | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
E2 is also required for long-term episomal maintenance of viral genomes within replicating cells.27,273,357,475,556 The critical role of E2 in viral DNA replication is not sufficient to support long-term maintenance of the viral genomes. Genome maintenance requires, in addition, cis minichromosome maintenance elements consisting of multiple E2 binding sites.475 In the presence of E2, plasmids containing viral E2 binding sites associate with mitotic chromosomes. Furthermore, the chromatin attachment function correlates perfectly with the stable episomal maintenance of the viral plasmids.273 E2 facilitates viral genome segregation by interacting simultaneously with condensed mitotic chromatin and viral genomes, linking the viral plasmids to the cellular mitotic chromosomes, and thereby ensuring the viral genomes are contained within the nuclear envelope when it reforms during telophase. The TA domain is required for the association of E2 with mitotic chromosomes and for the genome maintenance function in dividing cells. Specific mutations in the TA domain have been shown to disrupt the tethering of viral genomes to mitotic chromosomes.2,28,693 The cellular protein Brd4 mediates the association of BPV-1 E2 to mitotic chromosomes and may play an important role for some other PVs.688 Furthermore the binding of E2 to Brd4 is conserved among all the papillomaviruses.274,539,688
Additional cellular factors are likely to be involved in E2-mediated tethering to mitotic chromosomes and PV genome maintenance. For instance ChlR1 has been shown to be important for loading E2 onto mitotic chromosome.453 The E2 proteins of some of the alpha genus HPV types (HPV11, HPV16, and HPV18) have been shown to associate with the mitotic spindle rather than the chromosomes.629 It has been proposed that an association between mitotic spindles and these HPV E2 proteins might provide a mechanism for HPV viral genome persistence in host cells. Finally HPV8 E2 binds as large speckles at the ribosomal DNA loci at the pericentromeric regions of chromosomes.480 Therefore, different papillomavirus E2 proteins use different mechanisms to ensure the stable maintenance of their genomes in host cells and evaluation of these differences is likely to be fruitful.
The interaction of E2 with Brd4 is required for the transcriptional activation function of E2.539 Amino acid residues in the E2 TA domain required for Brd4 binding are also required for transcriptional activation, and siRNA knockdown of Brd4 protein levels reveals a role for Brd4 in E2 transcriptional activation.539 Brd4 is a member of the bromo and extra terminal (BET) family, a group of structurally related proteins characterized by the presence of two bromodomains and one extraterminal (ET) domain of unknown function.141 Bromodomains in general have been shown to interact with acetylated lysines in histones and are involved in chromatin targeting and remodeling.676 Unlike other bromodomain proteins, which are released from chromatin during mitosis, BET family members remain bound to chromatin during mitosis. Brd4 has been shown to influence the general RNA polymerase II–dependent transcription machinery by interacting with the core factors of the positive transcription elongation factor b (P-TEFb).281,684
Late Gene Expression
The viral late functions, such as vegetative viral DNA synthesis, capsid protein synthesis, and virion assembly, occur exclusively in differentiated keratinocytes. Transcriptional regulation of the late genes is directed from a specific promoter that becomes active only in terminally differentiated keratinocytes. The late genes include the capsid genes L1 and L2, as well as E4, which is located in the early region of the viral genomes. Of interest the late promoters for the human papillomaviruses that have been analyzed do not map to the LCR. Instead, a differentiation-specific promoter (referred to as P742 in HPV31 and P670 in HPV16) has been identified within the E7 coding region which gives rise to mRNAs that map heterogeneously over a 100-bp region in the E7 gene.221,266 Keratinocyte differentiation by itself is able to activate low levels of late transcription, and genome
amplification also increases the level of late gene expression.48,574 Recent studies have also established differentiation changes in the levels of the CCAAT/enhancer binding protein (C/EBP), β repressors and activators in the regulation of the HPV late promoter.230
amplification also increases the level of late gene expression.48,574 Recent studies have also established differentiation changes in the levels of the CCAAT/enhancer binding protein (C/EBP), β repressors and activators in the regulation of the HPV late promoter.230
Papillomavirus L1 and L2 gene expression is also regulated at a posttranscriptional level. Cis elements have been described for BPV1 as well as several HPV types that regulate late gene expression at a posttranscriptional level. In the 3′ untranslated regions (UTRs) of the late RNAs of each of these viruses there are negative regulatory elements that can inhibit the stability of late messenger RNAs. A negative regulatory element in the HPV16 3′ UTR contains multiple 5′ splice-like sequences, as well as an inhibitory GU-rich region that reduces mRNA stability and binds to specific cellular factors.142,305,322 Three different cellular factors (the U2 auxiliary splicing factor 65-kD subunit, the cleavage stimulation factor 64-kD subunit, and the Elav-like HuR protein) interact with the RNA and regulate its levels at a post-transcriptional level.322 In HPV1, an AU-rich inhibitory region has been identified in the 3′ UTR that also binds the Elav-like HuR protein.568 The model emerges that these, and perhaps additional cellular factors, are responsible for the nuclear retention or cytoplasmic instability of the nuclear retention element (NRE)-containing late transcripts. Keratinocyte differentiation would then lead to changes in these cell-encoded factors, thereby relieving the inhibition of late mRNA processing.
Virion Assembly and Release
Virion assembly takes place in the nuclei of terminally differentiated keratinocytes in which vegetative viral genome replication and expression of the virion proteins has occurred.152 Nuclear entry of L1 and L2 is mediated by cellular karyopherins, particularly the Kap alpha2/beta1 heterodimer.174,408 In addition to nuclear transport, karyopherin binding may prevent premature L1 assembly in the cytoplasm.43 As noted above, L1 can assemble into VLPs. However, L2 may increase the efficiency of the assembly reaction.313,694 Hsc70 may also participate in the assembly reaction, since it is found in association with nuclear L2 but is displaced in L1/L2 capsids that have packaged DNA.184 Packaging of the viral genome by the capsid proteins does not appear to require a sequence-specific packaging signal because many bacterial plasmids with no PV sequences can be efficiently packaged, at least in cultured cells, provided they are less than 8 kb in length.74 Preferential encapsidation of the viral genome may involve a size discrimination mechanism. Nascent capsids might randomly coalesce around any nuclear DNA but would generally form unstable open structures. A stable closed structure could assemble only if the DNA molecule is approximately the size of the 8-kb viral genome. Consistent with this hypothesis, linear fragments of cellular DNA less than 8 kb in length are efficiently encapsidated if the nuclei of L1/L2 expressing cells are gently lysed and exposed to a double-stranded DNA endonuclease.75 In cell culture systems, the L2 dependence for DNA encapsidation varies by PV type. For example, almost no DNA is encapsidated when BPV1 L1 is expressed alone, whereas HPV16 L1 alone rather efficiently encapsidates pseudoviral genomes or linear cellular DNA fragments, although the resulting DNA-containing L1-only capsids are essentially noninfectious.75
Upon exposure to an oxidizing environment, as occurs in the upper layers of a terminally differentiated squamous epithelium,114 the capsids are further stabilized by the formation of disulfide bonds between conserved cysteines on adjacent L1 monomers. This maturation process condenses the capsid and increases its regularity and resistance to proteolytic digestion.75 Formation of disulfide-linked L1 dimers and trimers was observed after capsid production in replicating cultured cells or in vitro raft cultures.75,115 Neither L2 nor encapsidated DNA appreciably influences the formation of L1 disulfide bonds. However, the extent of cross-linking varies by PV type, for example, being much greater for BPV1 than HPV16.75,668 PVs are not believed to be cytolytic, and release of the virions is thought to occur as a result of the normal loss of nuclear and cytoplasmic membrane integrity during terminal differentiation of the infected keratinocyte. E4-mediated collapse of cytokeratin filaments might assist in virion release.153
Viral DNA Replication
Little is known about the initial stages of viral DNA replication and amplification that occur following infection of a basal keratinocyte, when there is an amplification of the viral genome to approximately 50 to 100 copies. In cells in which the viral DNA has been established, the viral DNA is maintained as a stable multicopy plasmid. The viral genomes replicate an average of once per cell cycle during S-phase in synchrony with the host cell chromosome.207 As a multicopy plasmid, this type of DNA replication ensures a persistent infection in the basal cells of the epidermis. Vegetative DNA replication occurs in the more differentiated epithelial cells of the papilloma. Such differentiated cells have exited the cell cycle and are no longer capable of supporting cellular DNA synthesis. Through E6 and E7, however, the HPVs activate the DNA replication machinery to support vegetative viral DNA synthesis producing the genomes to be packaged into progeny virions.
Origin of DNA Replication
Papillomavirus DNA replication requires the origin of DNA replication in cis and the viral E1 and E2 proteins in trans. The minimal origin of DNA replication contains an A + T rich region (ATR), the E1 binding site that includes a region of dyad symmetry (DSR), and an E2 binding site.624 Origin-dependent DNA replication can be achieved in vitro with cell extracts containing high levels of E1 alone in the absence of E2.543,681 E1 is the essential virus-encoded replication factor and functions as an ATP-dependent helicase. The role of E2 is as an auxiliary factor in viral DNA replication; the binding of E1 to the origin of replication is stabilized through its interaction with E2 and the binding of E2 to its cognate sites adjacent to the origin.416,624
The E1 Protein
The E1 protein is highly conserved among the PVs, and the BPV1 E1 protein is a 68-kD nuclear phosphoprotein that binds specifically to the origin of replication.616,624,664 By itself, E1 binds the origin with weak affinity; however, this binding is stabilized through its interaction with E2. E1 has DNA-dependent ATPase and DNA helicase activities.65,543,682 E1 is required for both the initiation and elongation of viral DNA synthesis.368 In addition to its interaction with E2, E1 has been shown to bind a number of cellular proteins. E1 interacts with the p180 subunit of the cellular DNA polymerase α-primase and thereby recruits the cellular DNA replication initiation machinery to the viral replication origin.52,457 Several additional host
cellular proteins have been found to bind E1, including histone H1,596 SW1/SNF5,354 cyclin E/Cdk2,121,385 Hsp40/Hsp70,369 and Ubc9.492,685 Although the physiologic significance of some of these interactions remains to be determined, several appear to be quite interesting. In particular, the efficient cell cycle–regulated replication of papillomavirus genomes is dependent upon the association of E1 with the S-phase specific cyclin E-Cdk2 complex.121 In addition, the interaction of E1 with Ubc9 is required for efficient origin-dependent replication.685 E1 is small ubiquitin-like modifier 1 (SUMO-1) modified by Ubc-9, and this modification is required for the intranuclear accumulation of E1.493
cellular proteins have been found to bind E1, including histone H1,596 SW1/SNF5,354 cyclin E/Cdk2,121,385 Hsp40/Hsp70,369 and Ubc9.492,685 Although the physiologic significance of some of these interactions remains to be determined, several appear to be quite interesting. In particular, the efficient cell cycle–regulated replication of papillomavirus genomes is dependent upon the association of E1 with the S-phase specific cyclin E-Cdk2 complex.121 In addition, the interaction of E1 with Ubc9 is required for efficient origin-dependent replication.685 E1 is small ubiquitin-like modifier 1 (SUMO-1) modified by Ubc-9, and this modification is required for the intranuclear accumulation of E1.493
E2 Protein Replication Functions
Papillomavirus DNA replication requires E2 as an auxiliary factor.623,681 Although not essential for origin-dependent DNA replication in vitro, E2 greatly stimulates the ability of E1 to initiate DNA replication.681 E2 interacts with E146,383,416 and greatly enhances the ability of E1 to bind the replication origin.416,541,544 E2 can relieve nucleosome mediated repression of papillomavirus DNA replication in vitro.361 The E1–E2 complex is a precursor to a larger multimeric E1 complex, which after the removal of E2 can distort the replication origin and ultimately unwind the DNA (Fig. 54.10).384 E2 serves as an auxiliary factor that fosters the assembly of the preinitiation complex at the origin, but E2 itself plays no intrinsic role in viral DNA replication. A hexameric form of E1 protein is associated with the ATPase and DNA helicase activities intrinsic to its initiator function in DNA replication.186,542
Vegetative Viral DNA Replication
Vegetative replication of papillomavirus DNA is necessary to generate the genomes to be packaged in virions, a process that normally occurs only in the terminally differentiated epithelial cells of a papilloma. The mechanisms regulating the switch from plasmid maintenance to vegetative viral DNA replication are not known. The switch may involve the presence or absence of controlling cellular factors in differentiating keratinocytes. In addition, or alternatively, the relative levels of viral factors such as E1 or E2 (or their modification) may change in terminally differentiating keratinocytes. There have been few studies that have examined the mode of vegetative viral DNA replication in differentiated cells. One might anticipate that, as with the polyomaviruses, vegetative DNA replication occurs bidirectionally through theta structure intermediates, as it does in the maintenance replication phase. One intriguing study, however, suggests that there may be a switch from a bidirectional mode of replication to what could be a rolling circle mode.182 Additional studies on the mechanism of vegetative replication would appear to be warranted.
Viral Transformation
BPV-1 Transformation
Certain papillomaviruses are capable of inducing cellular transformation in tissue culture. The best studied of the transforming papillomaviruses is BPV1. Morphologic transformation in tissue culture was first described for BPV in the early 1960s.45,50,612 In the late 1970s, a focus assay was developed using established cell lines to study BPV1 transformation.163 In general, investigators have relied upon mouse C127 cells and NIH 3T3 cells for these transformation studies, although a variety of other rodent cells, including hamster and rat cells, are susceptible to BPV1–mediated transformation. Transformation of mouse C127 cells by BPV1 causes alterations in morphology, loss of contact inhibition, anchorage independence, and tumorigenicity in nude mice.163
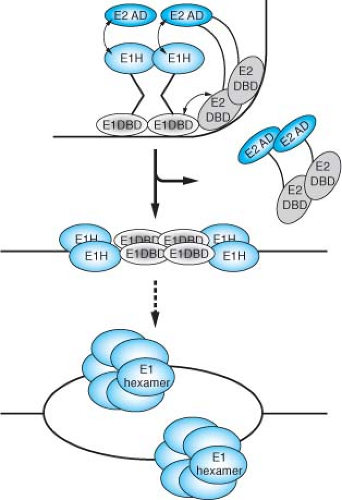 Figure 54.10. Proposed pathway for the assembly of an initiation-competent complex at the bovine papillomavirus (BPV) origin of DNA replication. The E1 initiator binds cooperatively with E2 to the ori forming a specific E12E22–DNA complex. As a consequence of the interaction between the E1 and E2 DNA binding domains (DBDs), a sharp bend is induced in the ori DNA. The bend promotes the interaction between the E1 helicase domain (E1H) and the E2 transactivation domain (E2AD). The resulting highly sequence-specific complex serves to recognize the ori. In a reaction requiring ATP hydrolysis, E2 is displaced and additional E1 molecules are added to the complex, resulting in the formation of a complex where four molecules of E1 are bound to the ori. This complex can distort the DNA duplex to and give rise to partially single-stranded regions. Subsequently, additional molecules are added. In a final step, E1 is assembled onto the exposed single strands forming a hexameric ring-like structure that constitutes the replicative helicase. (Courtesy of Arne Stenlund; modified from Enemark E, Chen G, Vaughn DE, et al. Crystal structure of the DNA binding domain of the replication initiation protein E1 from papillomavirus. Molec Cell 2000;6:149–158.) |
One notable characteristic of BPV1 transformed rodent cells is that the viral DNA is maintained as a stable multicopy plasmid.349 Integration of the viral genome is not required for either the initiation or maintenance of the transformed state. However, transformation is dependent upon the continued expression of viral genes as evidenced by the loss of the transformed phenotype in mouse cells that have been “cured” of the viral DNA by treatment with interferon.622
Genetic studies mapped the BPV1 transforming genes to the E5, E6, and E7 ORFs. The E5 gene is the major transforming gene of BPV1 in transformed cells. E5 encodes a small (44 amino acid) integral membrane protein that is sufficient for the transformation of certain established rodent cells in culture, and does so by activating the platelet derived growth factor (PDGF) β receptor to transform cells in a ligand-independent manner.467,468 The molecular biology of BPV1 E5 and its mechanism of transformation are described in more detail in Chapter 7. E5 is highly conserved among the group of papillomaviruses that induce fibropapillomas in their natural host and have the capacity to induce fibroblastic tumors in hamsters. The E5 gene is believed to be responsible for the proliferation of dermal fibroblasts in fibropapillomas.
The E6 and E7 genes of all the papillomaviruses encode proteins with conserved structural motifs. They contain domains of almost identically spaced CYS-X-X-CYS motifs (four in E6 and two in the carboxy-terminal portion of E7). It has been postulated that the E6 and E7 genes may have arisen from duplication events involving a 39-codon core sequence containing one of these motifs.112 The CYS-X-X-CYS motifs found in a number of nucleic acid binding proteins are characteristic of zinc-binding proteins. The papillomavirus E6 and E7 proteins bind zinc through these cysteine residues.24,225 BPV1 E6 and E7 have not been studied as extensively as their HPV counterparts, and they appear to transform through p53- and pRB1-independent mechanisms. As such, studies on the mechanisms by which BPV1 E6 and E7 transform cells could provide insights into the p53 and pRB independent activities of the HPV E6 and E7 oncoproteins, discussed in detail below.
HPV Immortalization and Transformation
The HPV16 and HPV18 genomes are not as efficient as BPV1 DNA at inducing transformation of established rodent cells; however, transformation can be achieved when the HPV DNA is transfected along with a second selective marker, such as the neomycin resistance gene.686 Immortalization assays employing primary rodent cells, primary human fibroblasts, and/or primary human keratinocytes have proven more informative. In such assays, the high-risk HPVs, such as HPV16 and HPV18, are positive for immortalization or transformation, whereas the low-risk viruses, such as HPV6 and HPV11, are not.535,585 These assays permitted the mapping of the E6 and E7 as oncogenes for the high-risk HPV types.
In established rodent cells, such as the NIH3T3 cells, the E7 ORF scores as the major HPV transforming gene.471,605,640,647,687 HPV16 and HPV18 by themselves are not able to transform primary rat fibroblasts or baby rat kidney cells.343,471 However, the E7 gene can cooperate with an activated ras oncogene to fully transform primary rat cells.30,362,440,471,585
The DNAs of the high-risk HPVs can also be distinguished from the DNAs of the low-risk HPVs by their abilities to immortalize primary human fibroblasts, human foreskin keratinocytes, or human cervical epithelial cells.161,476,535,646,673 The resulting cell lines are neither anchorage-dependent nor tumorigenic in nude mice, but they do display altered growth properties and are resistant in the response to signals for terminal differentiation.161,302,476,535
HPV E6
The HPV E6 proteins are approximately 150 amino acids in size and contain four Cys-X-X-Cys motifs that are involved in binding zinc.24,225,226 The first transforming activity identified for the high-risk alpha genus HPV E6 proteins (such as HPV16 and HPV18) was the ability complement E7 in the immortalization of human keratinocytes.245,421 This activity was soon explained by its ability to complex p53,655 a property not possessed by the low-risk HPV E6 proteins. Through its interaction with p53, E6 blocks the transcriptional function of p53 to activate p53-responsive promoters.412 The protein levels of p53 are generally quite low in HPV-positive carcinoma cell lines and in cells immortalized by the HPV oncoproteins,523 due to the ability of the E6 proteins of high-risk HPV types to promote the ubiquitin-dependent degradation of p53.525 Expression of the high-risk E7 proteins and their engagement of the pRB family of proteins results in an increase in the levels of p53 within cells, which in turn transcriptionally activates the expression of cell cycle arrest genes or proapoptotic genes.287 Indeed the half-life of p53 is dramatically decreased in E6-expressing cells, and E6 prevents the increase in p53 levels when cells are challenged with genotoxic agents.260,523 In targeting p53, the high-risk HPV E6 proteins inhibit DNA damage and oncogene-mediated cell death signals.167,287,308 Therefore, like SV40 Tag and Ad E1B that also target p53, HPV E6 has antiapoptotic activities and can interfere with the cell cycle regulatory functions of p53. E6 also can induce genomic instability, as evidenced by the development of translocations and aneuploidy in culture,498,657 as well as maintenance of stable episomal replication during the viral life cycle,458 and immortalization of human mammary epithelial cells.23,549
HPV16 E6 induces p53 degradation by forming a complex with the cellular ubiquitin-protein ligase E6AP,263,264 which is then able to bind and ubiquitylate p53.522 E6AP is the founding member of a class of ubiquitin-protein ligases called HECT E3 proteins, which directly transfer ubiquitin to their substrates.524 The catalytic domain of HECT proteins is a conserved 350 amino acid region defined by its homology to the E6AP carboxy terminus (HECT).262 The HECT domain binds to specific E2 enzymes and contains an active site cysteine residue that forms a thioester bond with ubiquitin262,524 (Fig. 54.11). Structure studies have determined that the HECT domain is a bilobed structure, with a larger N-terminal lobe that interacts with the ubiquitin-conjugating enzyme, and a smaller C-terminal lobe containing the catalytic cysteine residue.257
Levels of p53 in E6-immortalized cells or in HPV-positive cervical carcinoma cells are, on average, two- to threefold lower compared to primary cells.523 In uninfected cells, intracellular p53 levels increase significantly in response to DNA damage or genotoxic stress.299 The higher levels of p53 can result in a G1 growth arrest or apoptosis, as part of a cell defense mechanism that allows for either the DNA damage to be repaired prior to the initiation of a new round of DNA replication or the removal of the cell. E6-expressing cells, however, do not manifest a p53-mediated cellular response to DNA damage,308 indicating the ability of E6 to promote the degradation of p53 and prevent the steady level of p53 to rise above a certain threshold level (Fig. 54.12). Under DNA-damaging conditions, the E6-stimulated degradation of p53 abrogates the negative growth regulatory effect of p53, and as such contributes to genomic instability. E6AP does not normally regulate p53 ubiquitylation in the absence of E6. In binding E6AP, E6 directs E6AP to p53 allowing it to form a ternary complex. It should be noted that E6AP does not regulate p53 protein
stability in non–E6-expressing cells31,602: the ubiquitin ligase MDM2 is the major E3 ubiquitin ligase responsible for p53 degradation in the absence of E6.244,334
stability in non–E6-expressing cells31,602: the ubiquitin ligase MDM2 is the major E3 ubiquitin ligase responsible for p53 degradation in the absence of E6.244,334
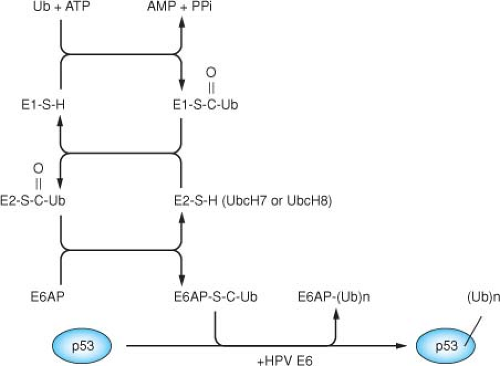 Figure 54.11. A ubiquitin thioester cascade model for the human papillomavirus (HPV) E6 dependent ubiquitination of p53. The E6 protein binds to the cellular protein E6AP, and the complex together functions as an E3 (ubiquitin protein ligase) in facilitating the ubiquitination of p53.522 The ubiquitination of a protein involves three cellular activities: E1 (ubiquitin activating enzyme), E2 (ubiquitin conjugating enzyme), and E3 (ubiquitin protein ligase). Ubiquitin is activated in an ATP-dependent manner and forms a high energy thioester with E1, which can then be transferred to the E2 through a thioester linkage. Ubiquitin can then be transferred to a cysteine within the Hect domain of E6AP, again as a thioester linkage524 through the direct binding of E6AP with UbcH7 or UbcH8.337 In conjunction with HPV-16 E6, E6AP then recognizes p53 and catalyzes the formation of an isopeptide bond between the carboxy-terminal glycine of ubiquitin and a lysine side chain of p53. In catalyzing the ubiquitination of p53, HPV-16 E6 also induces the self-ubiquitination and proteolysis of E6AP.295 |
The HPV16 E6 protein binds to E6AP within the N-terminal substrate recognition domain, directing E6AP to ubiquitylate p53.265 The E6AP protein is encoded by the UBE3A gene that is located in an imprinted region on chromosome 15q11-q13, and it has been linked to Angelman’s syndrome, a neurogenetic disorder characterized by severe mental retardation, ataxia, loss of speech, seizures, and other abnormalities.314,396 Several potential E6-independent substrates of E6AP have now been identified, including the human homolog of the yeast RAD23 protein involved in nucleotide excision repair (HHR23A), the src-family kinase Blk, and the MCM7 subunit of replication licensing factor.335,338,442 In addition, E6 induces self-ubiquitylation of E6AP.295 It is conceivable that the redirection of E6AP activity toward p53 by E6 might affect (either enhance or inhibit) the targeting of the normal substrates of E6AP, and that such an alteration of E6AP activity could account for some of the transforming activity of E6. E6AP is a component of a number of cellular complexes, including the proteasome and a 2MDa complex that contains Herc2 and Neurl4.393 E6 is recruited to each of these complexes by E6AP. E6 encodes a number of p53-independent functions that are relevant to cellular transformation and immortalization, and there are HPV16 E6 mutations that separate p53 degradation from cellular immortalization.315,372 It is possible that some of the p53-independent activities of E6 are still mediated by E6AP through the activities of these other cellular complexes.
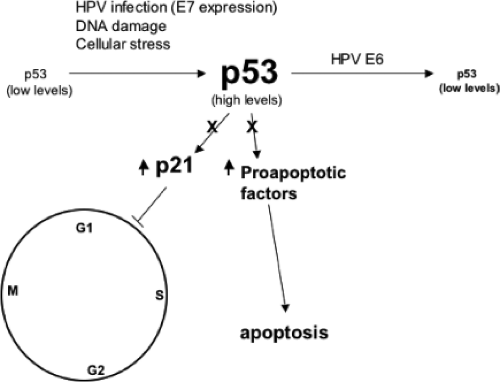 Figure 54.12. The level of p53 in primary cells is generally low. DNA damaging agents, viral infection, and expression of E7 increase the level of p53. Elevated levels of p53 can lead to either apoptosis or a cell-cycle checkpoint arrest in G1 through the transcriptional activation of proapoptotic genes or p21cip1. Viral oncoproteins may interfere with this negative growth regulatory function of p53, either by sequestering p53 into a stable, but nonfunctional complex (such as with SV40 TAg or the Ad5 55 kD E1B protein), or by ubiquitylation and enhanced proteolysis as observed with the high risk human papillomavirus (HPV) E6 proteins. |
A number of additional cellular targets have been identified for the high-risk alpha genus HPV E6 proteins (Table 54.2). Of note, the high-risk E6 oncoproteins contain an X-(S/T)-X-(V/I/L)-COOH motif at the extreme C-terminus that mediates binding with cellular PDZ domain–containing proteins. This motif
is unique in the high-risk HPV E6 proteins and is not present in the E6 proteins of the low-risk alpha genus HPV types. E6 serves as a molecular bridge between these PDZ domain proteins and E6AP, facilitating their ubiquitylation and mediating their proteolysis. Among the PDZ domain proteins implicated as E6 targets are hDlg, the human homolog of the Drosophila melanogaster discs large tumor suppressor, and hScrib, the human homolog of the Drosophila scribble tumor suppressor.197,427 Additional PDZ domain proteins shown to be capable of binding to E6 are the membrane associated guanylate kinase protein 1, 2 and 3 (MAGI 1, MAGI 2 and MAGI 3), the multi-PDZ domain protein (MUPP1) and the a cytoplasmic interacting protein containing a PDZ domain (TIP 2/GIPC).173,215,356,613 Several of the PDZ-containing proteins have been shown to be involved in negatively regulating cellular proliferation. Therefore some of the p53-independent transforming activities of the high-risk E6 oncoproteins may be linked to their ability to bind and degrade some of these PDZ motif–containing proteins.
is unique in the high-risk HPV E6 proteins and is not present in the E6 proteins of the low-risk alpha genus HPV types. E6 serves as a molecular bridge between these PDZ domain proteins and E6AP, facilitating their ubiquitylation and mediating their proteolysis. Among the PDZ domain proteins implicated as E6 targets are hDlg, the human homolog of the Drosophila melanogaster discs large tumor suppressor, and hScrib, the human homolog of the Drosophila scribble tumor suppressor.197,427 Additional PDZ domain proteins shown to be capable of binding to E6 are the membrane associated guanylate kinase protein 1, 2 and 3 (MAGI 1, MAGI 2 and MAGI 3), the multi-PDZ domain protein (MUPP1) and the a cytoplasmic interacting protein containing a PDZ domain (TIP 2/GIPC).173,215,356,613 Several of the PDZ-containing proteins have been shown to be involved in negatively regulating cellular proliferation. Therefore some of the p53-independent transforming activities of the high-risk E6 oncoproteins may be linked to their ability to bind and degrade some of these PDZ motif–containing proteins.
Table 54.2 Cellular Targets of the Papillomavirus E6 Oncoproteins | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
An important p53-independent activity of HPV16 E6 is its ability to activate telomerase in keratinocytes,320 through the transcriptional upregulation of the rate-limiting catalytic subunit of human telomerase (hTERT).315,443,632 Maintenance of telomere length is an important step in cellular immortalization and transformation, which occurs either through transcriptional activation of hTERT expression or through the activation of the ALT recombination pathway. Activation of hTERT is observed in most human cancers, including HPV-positive cervical cancers. The mechanism of hTERT promoter activation by E6 is complex. It involves the c-Myc transcription factor that binds to the hTert promoter, and it involves the E6AP-dependent degradation of a transcriptional repressor of the hTERT promoter, NFX1-91.205,206 Interactions of E6 with E6AP as well as c-Myc have been shown to be important in the transcriptional activation of the hTERT promoter.370,371,633,679
HPV16 E6 has also been reported to bind the transcriptional co-activator p300/CBP, a target also of Ad E1A and SV40 large T antigen.462,697 This interaction is limited to E6 proteins of high-risk HPVs associated with cervical cancer that have the capacity to repress p53-dependent transcription. The repression of p53 transcriptional activity by targeting the p53 co-activator CBP/p300 provides a second mechanism which can inhibit p53. A subsequent study has shown that in vitro, E6 can inhibit p300-mediated acetylation on p53 and nucleosomal core histones.614 A variety of other E6 cellular targets have been identified (Table 54.2); however, the physiologic relevance to transformation or immortalization has not yet been elucidated. It is possible that the binding of E6 to some of these targets might contribute to the virus–host cell functions unrelated to cellular transformation.
Other p53-independent activities for the alpha genus HPV E6 proteins have been described in the literature, including the activation of cap-dependent translation.573 Recent reviews that provide more detail on these activities have been published.256,403,418
Most studies thus far on the molecular biology of the HPV E6 proteins have been on the alpha genus high-risk HPV types. Studies of the cutaneous HPVs are more limited, and less is known about the cellular activities of the cutaneous HPVs of the beta genus. Recently, the beta HPV E6 types as well as BPV1 E6 have been shown to bind Mastermind-like 1 (MAML1) and other members of the Notch transcription complex.70,603 MAML1 is a core component of the transcriptional activation complex that mediates the effects of the canonical Notch signaling pathway.675 BPV1 and beta-HPV E6 repress Notch transcriptional activation, and this repression is dependent on an interaction with MAML1. Furthermore, the expression levels of endogenous Notch target genes are repressed by beta-HPV E6 proteins.603
Notch-dependent transcriptional programs are critical in the differentiation and cell cycle arrest of keratinocytes.378,491 In addition, inactivating Notch pathway mutations have been recently reported in squamous cell carcinomas of the head and
neck,3,588 and the skin,643 consistent with the notion that Notch signaling is a tumor suppressor pathway in squamous epithelial cells.155 E6 binding to MAML1 provides a novel mechanism of viral antagonism of HPV16 Notch signaling, and suggests that Notch signaling is an important epithelial cell pathway target for the beta-HPVs. Of interest, papillomavirus E6 proteins appear capable of binding to E6AP or to MAML1.
neck,3,588 and the skin,643 consistent with the notion that Notch signaling is a tumor suppressor pathway in squamous epithelial cells.155 E6 binding to MAML1 provides a novel mechanism of viral antagonism of HPV16 Notch signaling, and suggests that Notch signaling is an important epithelial cell pathway target for the beta-HPVs. Of interest, papillomavirus E6 proteins appear capable of binding to E6AP or to MAML1.
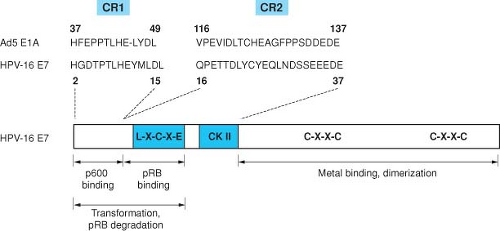 Figure 54.13. Amino acid sequence similarity between portions of conserved regions 1 and 2 (CR1 and CR2) of the Ad5 E1A proteins and the amino terminal 38 amino acids of HPV16 E7. CR2 contains the pRB binding site and the casein kinase II (CKII) phosphorylation site of HPV16 E7. |
HPV E7
The E7 protein encoded by the “high risk” HPVs is a small protein of about 100 amino acids, has been shown to bind zinc, and is phosphorylated by casein kinase II (CK II).402 E7 is a multifunctional protein that shares some functional similarities with adenovirus (Ad) 12S E1A.471 The HPV proteins also share important amino acid sequence similarity with portions of the AdE1A proteins and the SV40 large tumor antigen (TAg) (Fig. 54.13). These conserved regions are critical for the transforming activities in all three viral oncoproteins, and have been shown to participate in the binding to a number of important cellular regulatory proteins, including the product of the retinoblastoma tumor suppressor gene pRB, and the related pocket proteins, p107 and p130.135,165,659 Complex formation with pRB involves conserved region 2 of the Ad E1A protein and the corresponding region in the E7 protein and in SV40 large Tag.135,660
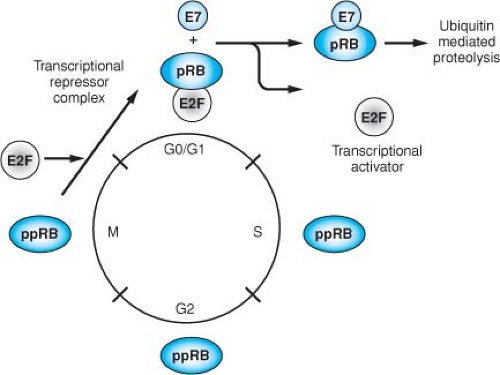 Figure 54.14. E7 abrogates the cell cycle regulation mediated by pRB (as well as the related proteins p107 and p130) by complex formation. During the cell cycle, pRB is differentially phosphorylated, and the underphosphorylated form is detected only in the G0/G1 phase. This underphosphorylated form is the active form of pRB, acting as a negative regulator of the cell cycle. During the transition to the S-phase, pRB is phosphorylated by cyclin-dependent kinases (cdk), resulting in the inactivation of its cell cycle regulatory functions. Members of the E2F family of cellular transcription factors are preferentially bound to the under-phosphorylated form of pRB, and in complex with pRB cannot activate transcription. Phosphorylation of pRB or complex formation with E7 results in the release of the E2F factors, allowing them to function as transcriptional activators of cellular genes involved in cellular DNA synthesis and progression into the S phase of the cell cycle. |
The retinoblastoma protein is a member of a family of cellular proteins that also includes p107 and p130, which are homologous in their binding “pockets” for E7, AdE1A, and SV40 TAg. Its phosphorylation state is regulated through the cell cycle, being hypophosphorylated in G0 and G1 and phosphorylated during S, G2, and M. pRB becomes phosphorylated at multiple serine residues by cyclin-dependent kinases at the G1/S boundary and remains phosphorylated until late M, when it becomes hypophosphorylated again through the action of a specific phosphatase (Fig. 54.14). The hypophosphorylated form represents the active form with respect to its ability to inhibit cell cycle progression. HPV16 E7, like SV40 TAg, binds preferentially to the hypophosphorylated form of pRB, resulting
in the functional inactivation of pRB through the release of E2F transcription factors, thus permitting progression of the cell into S phase of the cell cycle. This property of the viral oncoproteins to complex pRB accounts, at least in part, for their ability to induce DNA synthesis and cellular proliferation. The high-risk HPV E7 proteins associate with the pocket proteins and induce their proteasomal degradation.61,287 The LXCXE motif within the CR2 homology domain of E7 is sufficient for pocket protein binding,166,422 but additional sequences located in the immediate amino terminal CR1 homology domain of E7 are required for pocket protein degradation,287 and these sequences are also necessary for the transforming activities of E7.470
in the functional inactivation of pRB through the release of E2F transcription factors, thus permitting progression of the cell into S phase of the cell cycle. This property of the viral oncoproteins to complex pRB accounts, at least in part, for their ability to induce DNA synthesis and cellular proliferation. The high-risk HPV E7 proteins associate with the pocket proteins and induce their proteasomal degradation.61,287 The LXCXE motif within the CR2 homology domain of E7 is sufficient for pocket protein binding,166,422 but additional sequences located in the immediate amino terminal CR1 homology domain of E7 are required for pocket protein degradation,287 and these sequences are also necessary for the transforming activities of E7.470
Table 54.3 Cellular Targets of the Human Papillomavirus E7 Oncoproteins | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||
E7 targeting the pocket proteins, including pRB, is not sufficient to account for its immortalization and transforming functions, indicating that there are likely to be additional cellular targets of E7 that are relevant to cellular transformation.283 Table 54.3 provides a list of various cellular targets with which E7 has been shown to bind. The physiologic relevance of many of these interactions is unclear.
Of particular interest are the reports that E7 can interact with cyclin-dependent kinase (cdk) inhibitors. As with E1A, HPV16 E7 interacts with and abrogates the inhibitory activity of p27kip1.689 Because p27kip1 is involved in mediating cellular growth inhibition by transforming growth factor (TGF)-β in keratinocytes, this activity may contribute to the ability of E7 to override TGF-β–associated growth arrest.473 HPV16 E7 can also associate with p21cip1 and abrogate its inhibition of cdks as well as its inhibition of proliferating cell nuclear antigen (PCNA)-dependent DNA replication.194,286 p21cip1 is normally induced during keratinocyte differentiation,415 and presumably its inhibition by E7 may be critical to allowing the replication of papillomavirus DNA in differentiated squamous epithelial cells.101
E7 is necessary for the stable maintenance of HPV episomes in epithelial cells,181,610 and sequences in E7 that contribute to cellular transformation are also important for the functions in the viral life cycle.373,610 Hence, the ability of E7 proteins to induce DNA replication through the release of E2F transcription factor complexes and the inactivation of p21CIP1194,286 and p27KIP1689 is an essential component of the HPV replication strategy.
The high-risk HPV E7 proteins cause genomic instability in normal human cells.657 HPV16 E7 induces G1/S and mitotic cell cycle checkpoint defects and uncouples synthesis of centrosomes from the cell division cycle.420 This causes formation of abnormal multipolar mitoses, leading to chromosome mis-segregation and aneuploidy.157 Moreover, there is an increased incidence of double-stranded DNA breaks and anaphase bridges, suggesting that in addition to numerical abnormalities, high-risk E7 proteins also induce structural chromosome aberrations.158 Abnormal centrosome duplication rapidly results in genomic instability and aneuploidy, one of the hallmarks of a cancer cell. This activity is therefore likely to be functionally relevant to the contribution of high-risk HPV to malignant progression.
The HPV and BPV E7 proteins also bind to UBR4 (also known as p600).139,261 This binding appears to be conserved among all HPV E7 proteins (alpha as well as beta genera).658 The binding to p600 might therefore be important in mediating some of the pRB-independent functions of E7 such as modulating anoikis.138 UBR4 may be involved in N-end rule ubiquitylation, but its cellular function remains to be determined.
The high-risk HPV E7 proteins have also been shown to reprogram cellular transcriptional programs. This has been reviewed recently.403 E7 binds to both canonical and noncanonical E2F family members270,402 and affects their transcriptional activities. E7 binds E2F6, a component of the polycomb repressive complex, and the detection of E2F6/polycomb repressive complexes is decreased in E7-expressing cells.402
The repressive H3K27 marks, which are necessary for binding of polycomb repressive complexes, are decreased in HPV16 E7-expressing cells due to the transcriptional induction of the KDM6A and KDM6B H3K27-specific demethylases.401 HPV16 E7–mediated KDM6B induction accounts for expression of p16(INK4A).401 Moreover, KDM6A- and KDM6B-responsive Homeobox genes are expressed at significantly
higher levels, indicating that HPV16 E7 results in reprogramming of host epithelial cells. These effects are independent of the ability of E7 to inhibit the retinoblastoma tumor suppressor protein.
higher levels, indicating that HPV16 E7 results in reprogramming of host epithelial cells. These effects are independent of the ability of E7 to inhibit the retinoblastoma tumor suppressor protein.
What are the roles of the E6 and E7 oncoproteins in the normal life cycle of an HPV infection? It is likely that they function to allow the replication of the viral DNA. The viral E1 and E2 proteins are necessary for the initiation of viral DNA replication, but the virus is otherwise totally dependent on host cell factors, including DNA polymerase α, thymidine kinase, PCNA, and so on, for the replication of its DNA. These are proteins that are normally only expressed in S-phase during cellular DNA replication in cycling cells. Vegetative DNA replication for the papillomaviruses, however, occurs only in the more differentiated cells of the epithelium that are no longer cycling (see Fig. 54.5). Therefore, the papillomaviruses have evolved a mechanism similar to that of the polyomaviruses and the adenoviruses, to activate the cellular genes necessary for the replication of their own DNA in otherwise quiescent cells. These viruses may do so through the E7 proteins and their ability to release the E2F transcription factors by binding the pocket proteins including pRB (Fig. 54.13). In addition, E7 binds and inhibits the cdk inhibitor p21cip1 that is normally induced during keratinocyte differentiation, again presumably for the purpose of permitting viral DNA replication in a differentiated cell. The high-risk HPV E7 proteins, when expressed in the absence of E6, result in increased levels of p53 and in either a G1-mediated cell cycle arrest or apoptosis, depending upon the cell type. The mechanistic link between the pRB and p53 pathways is discussed in Chapter 7. E7 creates a signal that increases p53 levels. E6, by promoting the degradation of p53, counters this activity of E7 and permits the E7-dependent activation of the cellular DNA replication genes required for viral DNA replication.
HPV E5
Many of the papillomaviruses that induce purely epithelial papillomas (such as CRPV and the HPVs) contain E5 genes with the potential to encode short hydrophobic peptides. The structural similarity of these peptides to the BPV1 E5 protein has prompted studies of the potential transforming activities of the HPV E5 genes. The E5 proteins of the HPVs are required for optimal growth.175,201 In tissue culture, various HPV E5 genes have been shown to have some modest transforming activities, and in transgenic mice HPV16 E5 expressed in basal keratinocytes can alter the growth and differentiation of stratified epithelia and induce epithelial tumors at a high frequency.202
Although the biochemical mechanisms by which the E5 genes of the epitheliotropic papillomaviruses exert their growth stimulatory effects have not yet been fully elaborated, they may involve interactions with the epidermal growth factor receptor (EGFR) or the 16-kD subunit of the vacuolar ATPase, each of which has been shown to bind HPV E5 proteins.113,202,268,586,587,691 As with the BPV1 E5 protein, HPV16 E5 can bind the 16-kD subunit of the vacuolar ATPase and can inhibit the acidification of endosomes.113,502,586 It should be noted, however, that the E5 gene is not expressed in most HPV-positive cancers, suggesting that if the E5 gene does stimulate cell proliferation in vivo, it presumably functions in benign papillomas and not in the cancers. It might also participate in the initiation of the carcinogenic process or in some other aspects of the viral–host cell interaction relevant to the pathogenesis of the HPV infection. Indeed there are some data that would implicate E5 in the down-regulation of major histocompatibility complex (MHC) class II antigen expression.690
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


