As the drug development cycle moved into a new generation of anticonvulsants, it was suggested that MHD be developed in its own right as a new drug. This effort came to fruition in 2009 when the European Medicines Agency approved eslicarbazepine acetate (eslicarbazepine is (S)-(+)-MHD, also known as S-licarbazepine) for use as a prescription drug in the European Union. In 2013, a revised New Drug Application was submitted in the United States to the Food and Drug Administration, and it was approved in November of that year as a prescription-only agent for the treatment of partial seizures. The S-enantiomer was chosen as the sole component because it is the predominate isomer in the blood after oral administration of oxcarbazepine. Eslicarbazepine acetate is rapidly converted to eslicarbazepine, which, of course, has the same pharmacokinetic properties and pharmacodynamic effects as (S)-(+)-MHD produced after the administration of oxcarbazepine.
Because oxcarbazepine and eslicarbazepine acetate share an active moiety (known as either (S)-(+)-MHD, S-licarbazepine, or eslicarbazepine in various research studies), they are considered together in this chapter. Oxcarbazepine is covered in the first major section of the chapter, and eslicarbazepine is discussed in the second major section of the chapter. Because oxcarbazepine has been available for much longer period of time, it is to be expected that more is known about MHD after its administration compared to eslicarbazepine. But, in general, most of what is written about (S)-(+)-MHD in the oxcarbazepine section directly applies to eslicarbazepine.
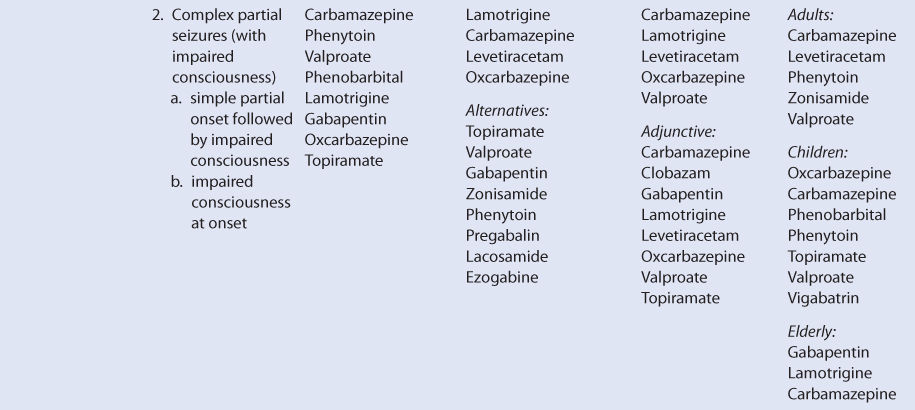
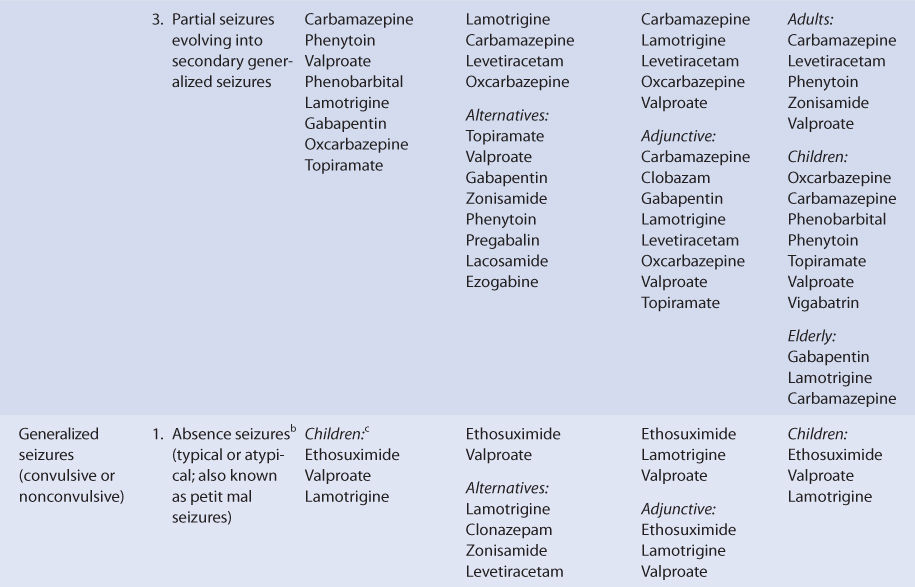
OXCARBAZEPINE
THERAPEUTIC AND TOXIC CONCENTRATIONS
Unlike most of the older antiepileptic drugs, the therapeutic range of oxcarbazepine is not well defined.12–14 However, serum concentration monitoring during oxcarbazepine treatment can be a useful adjunct in addition to monitoring treatment for therapeutic response and adverse effects. Although oxcarbazepine is measurable in the serum after oral administration of the drug, MHD (monohydroxy derivative), which is the active metabolite of oxcarbazepine, is the moiety that is actually monitored in clinical practice. After oral administration of oxcarbazepine, stereoisomers of MHD are produced by presystemic metabolism and additional hepatic clearance of oxcarbazepine in the ratio of about 4:1 [(S)-(+)-MHD:(R)-(–)-MHD].15–18 The therapeutic range for epilepsy used by many laboratories for MHD (MHD = (S)-(+)-MHD + (R)-(–)-MHD) is 3-35 μg/mL. Some patients will experience a higher incidence of adverse effects at concentrations in the upper end of the therapeutic range, so the efficacy of lower MHD serum concentrations should be assessed before moving to higher concentrations.12–14 The most common dose- or concentration-dependent side effects of oxcarbazepine are sedation, somnolence, fatigue, dizziness, ataxia, nausea, and headache.11,19
CLINICAL MONITORING PARAMETERS
The goal of therapy with anticonvulsants is to reduce seizure frequency and maximize quality of life with a minimum of adverse drug effects. While it is desirable to entirely abolish all seizure episodes, it may not be possible to accomplish this in many patients. Patients should be monitored for concentration-related side effects of oxcarbazepine therapy (sedation, somnolence, fatigue, dizziness, ataxia, nausea, headache). Other potential adverse reactions include diplopia, abnormal vision, abdominal pain, vomiting, dyspepsia, tremor, abnormal gait, and suicidal ideation or behavior. Some patients may develop hyponatremia during chronic therapy with oxcarbazepine, and serum sodium concentrations can be periodically measured to detect this problem.11,19–23
Severe allergic reactions, including anaphylaxis and angioedema, have been reported both after the first dose and after multiple doses of oxcarbazepine. Skin rashes can occur during oxcarbazepine therapy, and about 25%-30% of patients who developed a rash with carbamazepine will also develop a rash during oxcarbazepine treatment. Stevens-Johnson syndrome and toxic epidermal necrolysis have both been reported during oxcarbazepine therapy. Generally, these severe dermatologic adverse effects occur within 19 days of starting treatment. The incidence of these serious skin rashes is 0.5-6 cases per million-patient years. Should either of these dermatologic reactions develop, oxcarbazepine should be immediately discontinued and not restarted.11,19,23
Because epilepsy is an episodic disease state, patients do not experience seizures continuously. Thus, during dosage titration it is difficult to tell if the patient is responding to drug therapy or simply is not experiencing any abnormal central nervous system discharges at that time. Similarly, bipolar disorder also occurs at irregular time intervals. Oxcarbazepine doses should be carefully titrated to individual patient response and tolerability. Serum concentrations of MHD can be measured in patients as an adjunct to therapy.12–14 Concentration monitoring is particularly useful if a therapeutic concentration has been established for a patient and a new clinical event (drug interaction, new disease state or condition, etc) changes the pharmacokinetics of the drug. MHD concentrations can also be measured to document patient adherence to drug treatment or when a patient is switched between the oral dosage forms of oxcarbazepine to assure therapeutic levels are maintained.
BASIC CLINICAL PHARMACOKINETIC PARAMETERS
After oral administration, oxcarbazepine is extensively converted by presystemic metabolism and subsequent hepatic clearance via cytosol arylketone reductase to a monohydroxy derivative (MHD) in a stereospecific manner [(S)-(+)-MHD and (R)-(–)-MHD]. When oxcarbazepine is given orally, the ratio of (S)-(+)-MHD:(R)-(–)-MHD in the serum is about 4:1. MHD is further metabolized to glucuronide conjugates, which are principally eliminated by the kidney and to an inactive metabolite via oxidation that is a dihydroxy derivative known as DHD. Glucuronidation is undertaken by the UDP-glucuronosyltransferase (UGT) family of enzymes. After a single oral dose of oxcarbazepine, the urine contained the following compounds (expressed as percent of administered dose of oxcarbazepine): (S)-(+)-MHD = 22%, (S)-(+)-MHD-glucuronide = 39%, (R)-(–)-MHD = 5%, (R)-(–)-MHD-glucuronide = 6%, DHD = 3%, oxcarbazepine = undetectable.15–18 The range of typical half-life (t1/2) values for orally administered oxcarbazepine in adult subjects or patients with normal renal function and not taking enzyme inducers is 2.2-3.7 hours. For the same conditions, the range of typical half-lives is 13.0-17.0 hours for racemic MHD. For (S)-(+)-MHD and (R)-(–)-MHD, half-lives are 11.2 hours and 15.8 hours, respectively, after oral oxcarbazepine administration.15,17,24–26 Oxcarbazepine and (R)-(–)-MHD, but not (S)-(+)-MHD, appear to be substrates for P-glycoprotein transport using a mouse small intestine model.27
After intravenous administration of oxcarbazepine, presystemic metabolism obviously does not occur, and the drug is only eliminated by hepatic clearance. Since there is no hepatic first-pass effect for oxcarbazepine when given intravenously, the ratio of (S)-(+)-MHD:(R)-(–)-MHD in the serum is different and equals about 1.4:1. The distribution of compounds in the urine is also different after intravenous administration (expressed as percent of administered dose of oxcarbazepine): (S)-(+)-MHD = 16%, (S)-(+)-MHD-glucuronide = 32%, (R)-(–)-MHD = 12%, (R)-(–)-MHD-glucuronide = 13%, DHD = 4%, oxcarbazepine = undetectable. For (S)-(+)-MHD and (R)-(–)-MHD, half-lives are 10.6 hours and 9.0 hours, respectively, after intravenous administration of MHD.17
An injectable form of oxcarbazepine is not available for clinical use. For oral use, the drug is available as immediate-release tablets (150 mg, 300 mg, 600 mg), extended-release tablets (150 mg, 300 mg, 600 mg), and suspension (300 mg/5 mL). Oral bioavailability of oxcarbazepine is 100% using MHD concentrations as a comparator.17 The immediate-release dosage forms are rapidly absorbed from the gastrointestinal tract resulting in oxcarbazepine peak concentrations between 1.5 and 2 hours, MHD peak concentrations between 4 and 6 hours, and DHD peak concentrations 12-24 hours after a single dose of oxcarbazepine tablets.28–30 During multiple dose studies with oxcarbazepine tablets given every 12 hours, peak concentrations occur in about 1 hour for oxcarbazepine, in about 3 hours for MHD, and in about 4-6 hours for DHD.31,32 Peak concentrations of MHD after multiple doses of the extended-release tablet given once daily are observed about 7 hours after administration. During administration of multiple doses of 600 mg/d, the area under the concentration-time curve (AUC) and Cmax are approximately equivalent for immediate-release tablets given every 12 hours and extended-release tablets given once daily. But at a dosage rate of 1200 mg/d administered as multiple doses, the AUC and Cmax were about 19% lower for the extended-release tablets compared to the immediate-release tablets. Giving the extended-release tablet with meals, especially meals with a high fat content, can result in higher Cmax concentrations than a comparable dose of immediate-release tablets. Because of this, extended-release tablets should only be taken on an empty stomach (at least 1 hour before or at least 2 hours after meals).33,34 Rectal administration of an extemporaneously compounded oxcarbazepine fatty suppository or of oxcarbazepine oral suspension results in subtherapeutic MHD concentrations, so this route of administration with these dosage forms is not recommended.35,36
The AUC for MHD is proportional to the dose of oxcarbazepine that is administered, so doubling the dose of oxcarbazepine will double the AUC of MHD for a patient. In this sense, oxcarbazepine follows linear pharmacokinetics.25,26,37–39 The plasma protein binding of MHD is 35%-40%, and the average volume of distribution of MHD after intravenous administration in normal-weight adults is 47.9 L and 54.7 L for (S)-(+)-MHD and (R)-(–)-MHD, respectively.17,26,39–42 Oxcarbazepine is listed as a Category C medication using the FDA rating system for the use of drugs during pregnancy.
Oxcarbazepine, MHD, and DHD cross the placental barrier, and at birth the umbilical cord blood-to-maternal serum concentration ratio for each compound is approximately 1.0.43 Oxcarbazepine and MHD are transferred across the human placenta within about 15 minutes of maternal exposure, and placental tissue can convert oxcarbazepine into MHD.44 At birth, maternal and neonatal blood concentrations of both oxcarbazepine and MHD are equal. But after birth, serum concentrations of both compounds fell in a newborn who breastfed, so that 12% of the original concentration for oxcarbazepine (t1/2 = ∼22 hours) and 7% of the original concentration for MHD (t1/2 = ∼17 hours) were present by 5 days postpartum. The decline occurred despite continued consumption of oxcarbazepine by the mother. Oxcarbazepine and MHD cross into the breast milk of lactating mothers with a breast milk-to-maternal serum ratio of 0.50 for both agents.45
The blood/plasma, CSF/plasma, and saliva/plasma ratios for MHD during oxcarbazepine therapy are 1.3, 0.61, and 1.0, respectively.26,40,46,47 The concentration of MHD is about 14% lower in the human neocortex of the brain when compared to the concurrent serum concentration.19 Single-volume plasmapheresis conducted over a 3-hour time period removes 3%-4% of the daily oxcarbazepine dose and 5%-6% of the total body stores of MHD, so replacement doses of oxcarbazepine are not warranted.48 Oral activated charcoal (50 g, mixed with water) administered within 30 minutes of an oral oxcarbazepine dose decreased oxcarbazepine and MHD AUC values to 4% and 8%, respectively, of control values. Four repeated doses of oral activated charcoal (20 g, mixed with water) administered 12, 24, 36, and 48 hours after oxcarbazepine was given decreases MHD AUC and t1/2 values to 46% and 45%, respectively, of control values. Either of these modes of administration may be of use in acute overdose situations, depending upon the clinical scenario.49
EFFECTS OF DISEASE STATES AND CONDITIONS ON PHARMACOKINETICS AND DOSING
Assessing the effects of disease states and conditions on oxcarbazepine dosing and MHD pharmacokinetics is fraught with challenges. First and foremost is the obvious obstacle of the administration of a prodrug while tracking active metabolite concentrations and attempting to associate those concentrations to the ultimate therapeutic effect. Fortunately, the AUC for MHD is proportional to the dose of oxcarbazepine that is administered, so doubling the dose of oxcarbazepine will double the AUC of MHD for a patient. Using this framework, oxcarbazepine functionally follows linear pharmacokinetics with regard to MHD concentrations.25,26,37–39 However, the pharmacokinetic parameters for MHD are difficult to calculate because for most of the dosage interval MHD is being continually produced by metabolic conversion of oxcarbazepine. So, in most multiple dose cases, there is never a time during the dosage interval where there is a clear elimination phase for MHD. Especially for single dose studies done when oxcarbazepine was a new entity, serum concentrations for both oxcarbazepine and MHD were at or near the ability of the assay limits to detect the compounds, so accurate determination of pharmacokinetic parameters was difficult. Thus, it is unusual to have clearance, volume of distribution, or half-life values for MHD during oxcarbazepine administration. In most studies involving oxcarbazepine, only AUC, Cmax, Cmin, and Tmax are reported during multiple dose investigations. Finally, MHD is actually present in the body as the distinct stereoisomers, (S)-(+)-MHD and (R)-(–)-MHD, but most research articles simply report total MHD serum concentrations (total MHD = (S)-(+)-MHD + (R)-(–)-MHD. Due to these limitations, it is only possible to report semiquantitative findings in this section of the book.
The majority of a oxcarbazepine dose (60%-70%) is eliminated as MHD (30%-40%) or MHD-glucuronide (20%-40%) in the urine. Because of this, renal disease prolongs MHD half-life and decreases MHD clearance in a graded manner according to the degree of renal damage observed in the patient. Compared to adults with normal renal function (CrCl >90 mL/min), patients with a creatinine clearance (CrCl) of 30-80 mL/min have a 66% higher MHD AUC, patients with CrCl equal to 10-30 mL/min have a 147% higher MHD AUC, and patients with CrCl <10 mL/min have a 134% higher MHD AUC. The t1/2 of MHD progressively rises through the CrCl classifications (CrCl >90 mL/min, t1/2 = 10 hours; CrCl of 30-80 mL/min, t1/2 = 12 hours; CrCl of 10-30 mL/min, t1/2 = 16 hours; CrCl <10 mL/min, t1/2 = 19 hours). For MHD-glucuronide, the AUC values are 259%, 1392%, and 3512% higher than control for each of the respective CrCl categories. The AUC for oxcarbazepine also increases with decreasing renal function. While the prescribing information for oxcarbazepine recommends a 50% dosage decrease for patients with CrCl <30 mL/min and a slower titration rate, the results of this research study suggest that a 40% decrease in dose for patients with a CrCl = 30-80 (especially in the CrCl = 30-50 mL/min range) and a 60% decrease in dose for patients with a CrCl = 10-30 mL/min may be appropriate. The large store of MHD-glucuronide in patients with renal dysfunction is worth noting because similar metabolites for other drugs have been known to regenerate the active drug via de-glucuronidation. Because of this, caution is advised for patients taking oxcarbazepine with CrCl <10 mL/min. For patients with renal dysfunction, oxcarbazepine doses should be titrated to clinical response, and measurement of MHD serum concentrations can be a helpful adjunct. Due to the variability in the pharmacokinetics for MHD, patients with renal disease should be closely monitored for adverse effects due to oxcarbazepine therapy.23,50
After oral administration, oxcarbazepine is extensively converted by presystemic metabolism and subsequent hepatic clearance via cytosol arylketone reductase in a stereospecific manner ((S)-(+)-MHD:(R)-(–)-MHD serum ratio about 4:1). After an intravenous administration of oxcarbazepine, presystemic metabolism obviously does not occur, and the drug is only metabolized by hepatic clearance. Since there is no first-pass effect when oxcarbazepine is given intravenously, the ratio of (S)-(+)-MHD:(R)-(–)-MHD in the serum is different and equals about 1.4:1. Glucuronidation of MHD is undertaken by the UDP-glucuronosyl transferase family of enzymes.15,17,24–26 Oxcarbazepine and (R)-(–)-MHD, but not (S)-(+)-MHD, appear to be substrates for P-glycoprotein transport using a mouse small intestine model.27
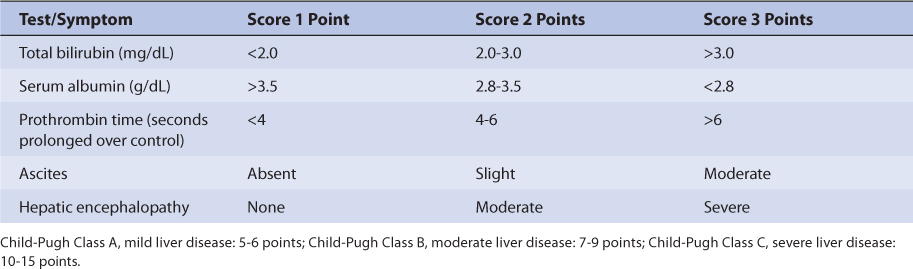
Based on this pattern of metabolism, one would expect liver dysfunction to alter oxcarbazepine and MHD pharmacokinetics. However, the only unpublished research article to evaluate oxcarbazepine therapy in patients with hepatic disease recommends no change in oxcarbazepine dosing in patients with mild or moderate hepatic dysfunction but did not include patients with severe liver disease.23,51 Because of this uncertainty for patients with hepatic dysfunction, oxcarbazepine doses should be titrated to clinical response, and measurement of MHD serum concentrations can be a helpful adjunct. Due to the variability in the pharmacokinetics for MHD, patients with hepatic disease should be closely monitored for adverse effects due to oxcarbazepine therapy. An index of liver dysfunction can be gained by applying the Child-Pugh clinical classification system to the patient (Table 17-2).52 Child-Pugh scores are completely discussed in Chapter 3 (Drug Dosing in Special Populations: Renal and Hepatic Disease, Dialysis, Heart Failure, Obesity, and Drug Interactions), but will be briefly discussed here. The Child-Pugh score consists of five laboratory tests or clinical symptoms: serum albumin, total bilirubin, prothrombin time, ascites, and hepatic encephalopathy. Each of these areas is given a score of 1 (normal) to 3 (severely abnormal; see Table 17-2), and the scores for the five areas are summed. The Child-Pugh score for a patient with normal liver function is 5 while the score for a patient with grossly abnormal serum albumin, total bilirubin, and prothrombin time values in addition to severe ascites and hepatic encephalopathy is 15.
TABLE 17-2 Child-Pugh Scores for Patients With Liver Disease
Adult and pediatric patients taking oxcarbazepine for the treatment of seizures have been studied while taking other antiepileptic drugs, and it is important to take concurrent medications into account when interpreting the findings of pharmacokinetic studies. Because the metabolism of oxcarbazepine does not include the cytochrome P-450 system, the induction (phenytoin, carbamazepine, phenobarbital, primidone) or inhibition (valproic acid and its derivatives) effects of other antiepileptics is moderated to some extent compared to other antiseizure medications. For adults, taking other antiepileptic drugs that are inducers simultaneously with oxcarbazepine decreases MHD AUC by about 30%-35% and decreases half-life by about 13%-18% for MHD. By comparison, taking valproic acid or one of its derivatives that are inhibitors concurrently with oxcarbazepine had little effect on oxcarbazepine or MHD concentrations.24,37 When lamotrigine was coadministered with oxcarbazepine, there were only slight decreases in oxcarbazepine and MHD AUC values (6-9%).32 For patients taking oxcarbazepine concurrently with valproic acid, there may be a small increase in the free fraction of MHD (64% for cotherapy, 57% for oxcarbazepine monotherapy), and there may be a slight impact of the volume of distribution for MHD in this situation.41 When other inducers or inhibitors are coadministered alone with oxcarbazepine, volume of distribution for MHD will likely remain unchanged due to the relatively low plasma protein binding for MHD.
For children taking oxcarbazepine, MHD clearance is about 50%-60% higher than in adults for the age range of 1 month to 3 years and is about 30%-160% higher than adults for older children. Pediatric patients taking other antiepileptic drugs that are inducers or inhibitors simultaneously with oxcarbazepine produces similar results for MHD pharmacokinetic parameters as in adults. The inducer-class antiseizure medications (phenytoin, carbamazepine, phenobarbital, primidone) increase clearance and decrease half-life for MHD compared to patients taking oxcarbazepine monotherapy. Less is known for the inhibitor-class antiseizure medications (valproic acid and its derivatives) in the pediatric population.53–56
For elderly men and women taking oxcarbazepine, the basic pattern compared with younger adults is similar to other antiepileptic drugs. For older women (age: 60-79 years), the AUC for MHD was 33% higher and t1/2 for MHD was 16% longer compared to younger women. Similarly, the AUC for MHD was 53% higher and t1/2 for MHD was 87% longer for older men (age: 61-82 years) compared to younger men. These results are consistent with the age-associated decreases measured in CrCl for the older population.23,57 These results suggest that initial doses for oxcarbazepine should be decreased by 33%-50% for elderly adults.
Using the FDA pregnancy classification system, oxcarbazepine is a Category C medication. Thus, oxcarbazepine is used to treat pregnant women when the potential benefits of drug therapy outweigh the potential risks. Pregnancy alters the steady-state concentrations of MHD in women taking oxcarbazepine. Compared to baseline values, MHD steady-state concentrations decrease during pregnancy by the following amounts: 72% during first trimester, 74% during second trimester, and 64% during third trimester. Postpartum, MHD steady-state concentrations return to baseline values (mean time 109 days postdelivery). Oxcarbazepine, MHD, and DHD cross the placental barrier, and at birth the umbilical cord blood-to-maternal serum concentration ratio for each compound is approximately 1.0.43 Oxcarbazepine and MHD are transferred across the human placenta within about 15 minutes of maternal exposure, and placental tissue can convert oxcarbazepine into MHD.44 At birth, maternal and neonatal blood concentrations of both oxcarbazepine and MHD are equal. But after birth, serum concentrations of both compounds fell in a newborn who were breastfed, so that 12% of the original concentration for oxcarbazepine (t1/2 = ∼22 hours) and 7% of the original concentration for MHD (t1/2 = ∼17 hours) were present by 5 days postpartum. The decline occurred despite continued consumption of oxcarbazepine by the mother.45 Oxcarbazepine and MHD cross into the breast milk of lactating mothers with a breast milk-to-maternal serum ratio of 0.50 for both agents.45
DRUG INTERACTIONS
After oral administration, oxcarbazepine is extensively converted by presystemic metabolism and subsequent hepatic clearance via cytosol arylketone reductase in a stereospecific manner (S)-(+)-MHD:(R)-(–)-MHD serum ratio about 4:1. After intravenous administration of oxcarbazepine, presystemic metabolism obviously does not occur, and the drug is metabolized solely by hepatic clearance. Since there is no first-pass effect when oxcarbazepine is given intravenously, the ratio of (S)-(+)-MHD:(R)-(–)-MHD in the serum is different than for orally administered oxcarbazepine and equals about 1.4:1. Glucuronidation of MHD is conducted by the UDP-glucuronosyl transferase family of enzymes.15,17,24–26 Additionally, oxcarbazepine and (R)-(–)-MHD, but not (S)-(+)-MHD, are substrates for P-glycoprotein transport using a mouse small intestine model.27 Because of this complex elimination pattern, there are ample opportunities for drug interactions.
Adult and pediatric patients taking oxcarbazepine for the treatment of seizures have been studied while taking other antiepileptic drugs. Because the metabolism of oxcarbazepine does not include the cytochrome P-450 system, the induction (phenytoin, carbamazepine, phenobarbital, primidone) or inhibition (valproic acid and its derivatives) effects of other antiepileptics is modulated to some extent compared to other anticonvulsants. For adults, taking other antiepileptic drugs that are inducers simultaneously with oxcarbazepine will decrease MHD AUC by about 30%-35% and decrease half-life by about 13%-18% for MHD. By comparison, taking valproic acid or one of its derivatives that are inhibitors concurrently with oxcarbazepine has little effect on oxcarbazepine or MHD concentrations.24,37,58 When lamotrigine was coadministered with oxcarbazepine, there were only slight decreases in oxcarbazepine and MHD AUC values (6%-9%).32 For patients taking oxcarbazepine concurrently with valproic acid, there may be a small increase in the free fraction of MHD (64% for cotherapy, 57% for oxcarbazepine monotherapy), and there may be a slight impact on the volume of distribution for MHD in this situation.41 When other inducers or inhibitors are coadministered alone with oxcarbazepine, volume of distribution for MHD will likely remain unchanged due to the relatively low plasma protein binding for MHD.
Pediatric patients taking other antiepileptic drugs that are inducers or inhibitors simultaneously with oxcarbazepine produces similar results for MHD pharmacokinetic parameters as in adults. The inducer-class antiseizure medications (phenytoin, carbamazepine, phenobarbital, primidone) increase clearance and decrease half-life for MHD compared to patients taking oxcarbazepine monotherapy. Less is known for the inhibitor-class antiseizure medications (valproic acid and its derivatives) in the pediatric population.53–56
Oxcarbazepine has the potential to alter the concentrations and pharmacokinetics of other drugs due to its ability to induce or inhibit the cytochrome P-450 system. Early in the development of oxcarbazepine, it was noted that oxcarbazepine 300 mg administered orally twice daily could increase the elimination of two marker agents, antipyrine and 6β-OH-cortosol, which are used as broad-based indicators of liver enzyme induction.59 At about the same time, a similar study that administered oxcarbazepine orally at a rate of 1500-2400 mg/d altered antipyrine clearance and half-life even more than lower doses of oxcarbazepine.60 Subsequent to these investigations, oxcarbazepine was found to be an inducer for CYP3A4.61 Oxcarbazepine therapy decreases the steady-state concentrations of many drugs, including ethinyl estradiol, levonorgestrel, rufinamide, lamotrigine, levetiracetam, pregabalin, and topiramate.32,62,67 In contrast to the effect on CYP3A4, oxcarbazepine and MHD are inhibitors of the CYP2C19 system and decrease the elimination of phenytoin.68
INITIAL DOSAGE DETERMINATION METHODS
Oxcarbazepine dosing is usually initiated using a “start low, go slow” type of titration process.11,19,23,34,69 This is a rational and preferred way to instigate dosing with the agent because the complex metabolic and elimination pattern yields widely different MHD concentrations depending on a variety of factors (patient age, concurrent antiepileptic therapy, etc), and it avoids the development of adverse effects early in treatment. Achieving a therapeutic effect without the development of side effects will improve patient adherence with oxcarbazepine therapy. There are several ways to start oxcarbazepine treatment, and one of the many recommended methods will be discussed in this section of the book as Literature-Based Recommended Dosing.11,19,23,34,69,70
For adults, oxcarbazepine treatment is usually started at a rate of 300-600 mg/d (immediate-release oral dosage forms given as equal, divided doses every 12 hours; extended-release tablets given once daily on an empty stomach). Every 1-2 weeks the dosage rate may be advanced by giving an additional 300-600 mg/d, and patients should be simultaneously monitored for seizure frequency and adverse drug effects. This dosage titration scheme can be used for adjunctive therapy with oxcarbazepine, oxcarbazepine monotherapy, or conversion from other antiepileptics to oxcarbazepine monotherapy. For patients that are undergoing conversion to oxcarbazepine monotherapy, the other anticonvulsants can be tapered off over a 3-6-week time frame, while the expected dose of oxcarbazepine is being ramped up during the first 2-4 weeks of this time period. The aim of this procedure is to ensure that effective amounts of oxcarbazepine will be prescribed early in the conversion process, before the other antiepileptic drugs are discontinued. Maximum tolerated doses of oxcarbazepine are usually in the 2400-3000 mg/d range.
For children, aged 4-16 years the initial dose of oxcarbazepine is 8-10 mg/kg/d for adjunctive therapy, conversion to oxcarbazepine monotherapy, or oxcarbazepine monotherapy. For adjunctive therapy with oxcarbazepine, the dose is slowly titrated to the recommended maximum over a 2-3-week time period (patient weight with maximum oxcarbazepine dosage: 20-29 kg, 900 mg/d; 29-39 kg, 1200 mg/d; ≥40 kg, 1800 mg/d). For conversion to oxcarbazepine monotherapy, oxcarbazepine doses are advanced at a rate of 10 mg/kg/d weekly, while the concomitantly administered antiepileptic therapy is tapered off over 3-6 weeks. For oxcarbazepine monotherapy, the dose is also slowly titrated, but at a rate of 5 mg/kg/d every 3-7 days. The maximum dose of oxcarbazepine monotherapy (either converted from other antiepileptic drugs or as initial therapy) is determined by patient weight: 900 mg/d for 20 kg, 1200 mg/d for 25-30 kg, 1500 mg/d for 35-45 kg, 1800 mg/d for 50-55 kg, 2100 mg/d for 60-70 kg. For younger children aged 2-3 years, the initial dose of oxcarbazepine for the adjunctive treatment is also 8-10 mg/kg/d, but if the patient weighs under 20 kg a dose of 16-20 mg/kg/d can be used. For younger patients, the dose of oxcarbazepine can be titrated over 2-4 weeks, and the dose should not exceed 60 mg/kg/d given in two divided doses.
For adult patients with severe renal dysfunction (CrCl <30 mL/min) the initial dose of oxcarbazepine should be reduced by ½ to 300 mg/d. Adults suffering from liver disease with mild to moderate hepatic impairment (see Table 17-2) can be started on the typical dose, but the dosage of oxcarbazepine for patients with severe liver dysfunction is unknown. Dosage adjustments for elderly patients should be made on the basis of their renal function (CrCl).
Literature-Based Recommended Dosing
EXAMPLE 1 
SO is a 51-year-old, 75-kg (height = 5 ft 10 in) male with complex partial seizures who requires adjunctive therapy with oxcarbazepine. He has a normal liver and renal function and is currently taking phenytoin (steady-state concentration = 18.2 μg/mL). Additional dosage increases have been tried with phenytoin, but they have been unsuccessful due to development of adverse effects. Suggest an initial oxcarbazepine dosage regimen using immediate-release tablets for this patient.
1. Estimate oxcarbazepine dose according to age, disease states and conditions, dosage form, and concurrent drug therapy.
For an adult patient taking phenytoin and treated with immediate-release oxcarbazepine tablets, the initial dose is 300-600 mg/d given as a divided dose every 12 hours.
For the first 2 weeks, the initial dose of 150 mg every 12 hours was chosen. Every 1-2 weeks of therapy, the dose can be escalated by an additional 300-600 mg/d to a maximum of 2400-3000 mg/d. The patient should be closely monitored for seizure frequency and adverse drug effects.
SO was titrated to a dose of oxcarbazepine 1200 mg every 12 hours without any additional side effects and a decreased seizure frequency.

SO is a 51-year-old, 75-kg (height = 5 ft 10 in) male with complex partial seizures who requires monotherapy with oxcarbazepine. He has normal liver and renal function. Suggest an initial oxcarbazepine dosage regimen using immediate-release tablets for this patient. (Note: This is a similar patient profile as Example 1, except for no concurrent antiepileptic drug therapy, so that the differences can be contrasted.)
1. Estimate oxcarbazepine dose according to age, disease states and conditions, dosage form, and concurrent drug therapy.
For an adult patient taking oxcarbazepine as monotherapy using immediate-release tablets, the initial dose is 300-600 mg/d given as a divided dose every 12 hours.
For the first 2 weeks, the initial dose of 300 mg every 12 hours was chosen. Every 1-2 weeks of therapy, the dose can be escalated by an additional 300-600 mg/d to a maximum of 2400-3000 mg/d. The patient should be closely monitored for seizure frequency and adverse drug effects.
SO was titrated to a dose of oxcarbazepine 1500 mg every 12 hours without any side effects and a decreased seizure frequency.
EXAMPLE 3 
SO is a 51-year-old, 75-kg (height = 5 ft 10 in) male with complex partial seizures who requires monotherapy with oxcarbazepine. He has normal liver and renal function. Suggest an initial oxcarbazepine dosage regimen using extended-release tablets for this patient. (Note: This is a similar patient profile as Example 2, except for use of extended-release tablets, so that the differences can be contrasted.)
1. Estimate oxcarbazepine dose according to age, disease states and conditions, dosage form, and concurrent drug therapy.
For an adult patient taking oxcarbazepine as monotherapy using extended-release tablets, the initial dose is 300-600 mg/d given once daily on an empty stomach.
For the first 2 weeks, the initial dose of 600 mg daily was chosen. Every 1-2 weeks of therapy, the dose can be escalated by an additional 300-600 mg/d to a maximum of 2400-3000 mg/d. The patient should be closely monitored for seizure frequency and adverse drug effects.
SO was titrated to a dose of oxcarbazepine 1800 mg daily without any side effects and a decreased seizure frequency.
EXAMPLE 4 
JX is a 9-year-old 35-kg (height = 4 ft 6 in) female with simple partial seizures who requires adjunctive therapy with oxcarbazepine. She has normal liver and renal function and is currently taking valproate (steady-state concentration = 81.7 μg/mL). Additional dosage increases have been tried with valproate, but they have been unsuccessful due to development of adverse effects. Suggest an initial oxcarbazepine dosage regimen using oral suspension for this patient.
1. Estimate oxcarbazepine dose according to age, disease states and conditions, dosage form, and concurrent drug therapy.
For a pediatric patient taking valproate and treated with oxcarbazepine oral suspension, the initial dose is 8-10 mg/kg/d given as two divided doses every 12 hours.
For the first 1-2 weeks, the initial dose of 150 mg every 12 hours was chosen (35 kg • 9 mg/kg/d = 315 mg, rounded to 300 mg/d). Every 1-2 weeks of therapy, the dose can be escalated by an additional 150-300 mg/d to a target dose of 1200 mg/d. The patient should be closely monitored for seizure frequency and adverse drug effects.
SO was titrated to a dose of oxcarbazepine 600 mg every 12 hours without any side effects and a decreased seizure frequency. Within 6 months, she was switched to oxcarbazepine tablets 600 mg every 12 hours when she could swallow whole tablets without any distress. Therapeutic response remained constant after the dosage form switch, with no change in seizure frequency or adverse effects.
USE OF MHD SERUM CONCENTRATIONS TO ALTER DOSES OF OXCARBAZEPINE
A definitive therapeutic range for steady-state MHD serum concentrations during oxcarbazepine treatment has not been established.12–14 Because of this, serum concentration monitoring for oxcarbazepine plays only a supportive role in the dosing of the drug. Important patient parameters (seizure frequency or frequency/severity of mood episodes, potential oxcarbazepine side effects, etc) should be followed to confirm that the patient is responding to treatment and not developing adverse drug reactions.8,11 However, there are clinical situations where MHD steady-state serum concentration monitoring of trough levels can be very helpful.
Once dosage titration has been conducted, and an effective dose of oxcarbazepine has been established by assessing therapeutic response and absence of adverse effects, many clinicians obtain a steady-state MHD concentration to establish an individualized effective target value for the patient. When this is done, if seizure frequency changes or an adverse effect occurs, another MHD serum concentration can be measured to ascertain potential solutions to the problem. For instance, if a patient suddenly has more seizures than usual, comparing the current MHD serum concentration with the established effective target MHD concentration can be helpful to decide a course of action. If the current MHD concentration is lower than usual, reasons for the change can be explored (lack of adherence to therapy, drug interaction, etc), and, if appropriate, an oxcarbazepine dosage increase can be considered to reestablish effective concentrations. If the current MHD concentration is about the same, changes to therapy may be warranted (increase oxcarbazepine dose, addition of other drug therapy, etc). Alternatively, if a patient suddenly develops adverse effects that could be due to oxcarbazepine therapy, and the current MHD serum concentration is high, it may be appropriate to decrease the oxcarbazepine dose. Also, when an effective MHD concentration has been established for a patient, the effects of new drug therapy that may cause drug interactions or of new disease states or conditions that can alter oxcarbazepine or MHD pharmacokinetics can be assessed prospectively by measuring the current MHD concentration before any clinical events occur.
Because a definitive therapeutic range for MHD has not been firmly established, and a “start low, go slow” dosage titration strategy has been established for the drug, oxcarbazepine dose increases are usually capped at the rate noted in the initial dosing section. When needed, dosage decreases are usually made using one of the smaller changes possible from available tablet strengths. Thus, pharmacokinetic calculations are usually made to compute the expected steady-state concentration after a specific oxcarbazepine dosage change has been determined rather than computing a new dose based on a desired target concentration. Because MHD follows linear pharmacokinetics during oxcarbazepine treatment, the pharmacokinetic calculations involved during dosage titration are straightforward.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


