- Linezolid is the first in a new class of antibacterial agents active against Gram positive bacteria. It has excellent bioavailability and distribution into body tissues, making it particularly useful for complicated skin and soft tissue infections and pneumonia caused by susceptible organisms.
- Resistance is not yet a major problem due to its restricted use.
- For further information, see Leach et al.(2011).
The only oxazolidinone antibiotic currently approved for clinical use is linezolid (Figure 4.5.1), which is indicated for the treatment of severe infections due to Gram positive organisms, as shown in Table 4.5.1.
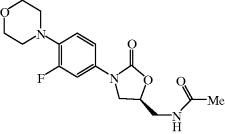
Table 4.5.1 Therapeutic indications for linezolid.
| Indication | Notes |
| Treatment of community-acquired pneumonia and nosocomial pneumonia | Only used when known or suspected to be caused by susceptible Gram positive bacteria |
| Complicated skin and soft-tissue infections | Only used when microbiological testing has established that the infection is known to be caused by susceptible Gram positive bacteria |
4.5.1 Discovery
The discovery of linezolid is an example of a careful medicinal chemistry approach to novel lead compound development, coupled with industrial competition and serendipity.
This interesting story started at E. I. duPont de Nemours & Company, during a screening programme in the 1980s, with the discovery of oxazolidinone-based molecules that displayed potent activity against Gram positive bacteria, even those known to be resistant to penicillins. DuPont announced their discovery with a series of articles between 1987 and the early 1990s (Brickner et al., 2008), having patented the oxazolidinones as antibacterial agents (Gregory, 1987). The lead compounds, DuP-105 and DuP-721 (Figure 4.5.2), were comparable in potency to vancomycin (Maple et al., 1989) and were found to have excellent activity against a range of streptococcal and staphylococcal strains (Leach et al., 2011). Within 2 years, scientists at the Upjohn Company (now part of Pfizer Inc.) had developed their own oxazolidinone series and had compounds that matched, or even improved upon, the activity of the DuPont lead compounds. The race to find the best antibacterial oxazolidinone was on! Around this time, DuPont received results from preclinical in vivo trials with their lead compound (DuP-721), which identified serious toxicity issues, and they subsequently abandoned their antibacterial oxazolidinone development programme. There must have been a sharp intake of breath at Upjohn when they heard this news and they rapidly carried out their own preclinical in vivo screen on the racemic form of their lead compound A, comparing it to the racemate of DuP-721, and found that lead compound A was at least as efficacious as the DuPont compound, but, amazingly, produced no toxicity problems. It seems remarkable that compound A could have such a different in vivo toxicology profile, as the structural differences are very small (Figure 4.5.2), so Lady Luck had smiled on the Upjohn team.
Figure 4.5.2 Structures of oxazolidinone lead and clinical candidates (Ford et al., 1997; Brickner et al., 2008). The oxazolidinone core is highlighted in each
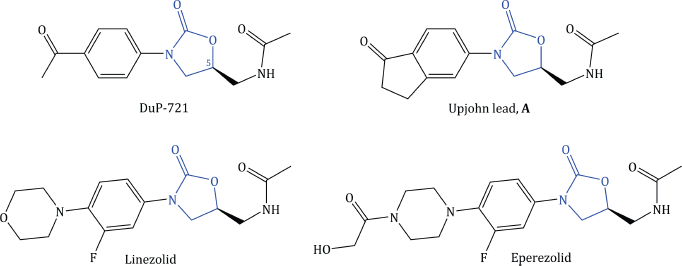
Things did not go entirely smoothly for the Upjohn team, though, as they soon found their lead compound A was already included in the patent filed by DuPont. Further careful work followed, which allowed initial structure and toxicology activity relationships (discussed further in Subsection 4.5.4) to be derived, and eventually resulted in the proposal of two clinical candidates, linezolid and eperezolid (Figure 4.5.3) (Ford et al., 1997; Brickner et al., 2008).
Figure 4.5.3 Early structure–activity relationship (SAR) for the oxazolidinones, which led to the development of linezolid and eperezolid (Barbachyn and Ford, 2003; Livermore, 2003; Brickner et al., 2008)
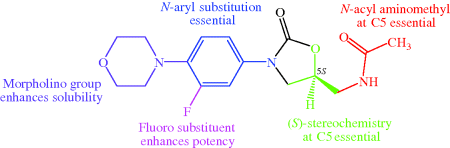
Both linezolid and eperezolid progressed into preclinical trials and were found to have very similar antibacterial activities. As is often seen in drug development, the choice between these similar drug candidates came down to which had the better pharmacokinetic profile.
There has also been controversy associated with the discovery of linezolid, this time with allegations of illegal marketing activities. The pharmaceutical companies Pharmacia AB and the Upjohn Company merged in 1995, and then merged with Pfizer Inc. in 2003, thus placing linezolid into Pfizer’s drug portfolio. In 2009, Pfizer agreed to pay around $2.3 bn (or £1.4 bn), ‘to settle civil and criminal allegations’ of having illegally promoted four unrelated drugs, including linezolid, for uses that did not have regulatory (FDA) approval. This must seem like an incredible fine, but was reported to amount to less than 3 weeks of Pfizer’s sales, reflecting the huge sums of money involved in the pharmaceutical industry (http://www.nytimes.com/2009/09/03/business/03health.html, last accessed 9 March 2012). For more on the discovery of linezolid, see Brickner et al. (2008).
4.5.2 Synthesis
Like that of chloramphenicol, the synthesis of linezolid and its analogues is relatively simple.22 The requirement for the (S)-stereochemistry at C5 was established early in the lead development of the oxazolidinones by DuPont and was easily solved by the use of a commercially available, enantiomerically pure, starting material.
There is more than one synthetic route to linezolid, but we will consider the initial industrial synthesis here (Scheme 4.5.1), which resulted from the optimisation of the chemistry at Upjohn and Pharmacia (Pearlman et al., 1998; Brickner et al., 2008). One drawback of this route is the need for expensive reagents, such as palladium on charcoal, which may have contributed to the relatively high cost of linezolid (currently estimated at about £900 for a course of 20 tablets of 600 mg each). Despite these costs, the pharmacoeconomic considerations in several countries, taking account of drug and hospitalisation costs, generally support the use of linezolid for its licensed applications.
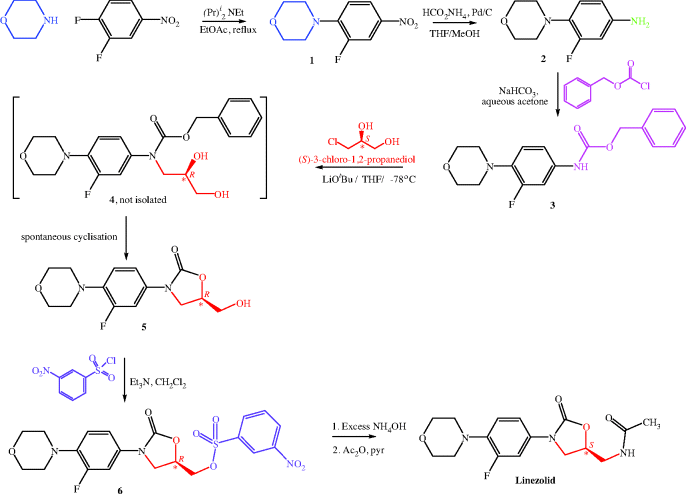
The first three steps of the synthesis shown in Scheme 4.5.1 are routine but effective, starting with a nucleophilic aromatic substitution of the 4-fluoro group in 3,4-difluoronitrobenzene by morpholine, followed by palladium-catalysed reduction of the aromatic nitro group in 1 to give the corresponding aromatic amine 2. Addition of a carbobenzyloxy (CBZ) group to the amine using benzylchloroformate produces the carbamate 3, which is deprotonated by lithium tertiary-butoxide (a strong base) and reacted with the commercially sourced chiral reagent (S)-3-chloro-1,2-propanediol. The intermediate 4 is not isolated, but spontaneously cyclises, with loss of benzyl alcohol, to form the five-membered oxazolidinone ring of 5, which reacts with 3-nitrophenylsulfonyl chloride to form the sulfonate ester 6. A one-pot nucleophilic substitution of the sulfonate ester with ammonia, released from ammonium hydroxide, followed by acetylation, forms linezolid in 65% overall yield (Brickner et al., 2008).
Although you may at first be concerned that the stereochemistry swaps from S in (S)-3-chloro-1,2-propanediol to R in intermediate 4, oxazolidinone 5, and sulfonate ester 6, then back to S in linezolid again, with no reactions at the chiral carbon (highlighted with a *), closer inspection will show you that it is due to the higher priority of a chlorine atom over an oxygen atom (or an oxygen atom over a nitrogen atom) when assigning the stereochemistry. The absolute configuration at the chiral carbon does not change throughout the reaction sequence; it is the prioritisation of the groups around the chiral carbon that changes (according to the presence of Cl, O, or N around the chiral carbon).
Many other oxazolidinones have been synthesised and tested for their antibacterial activity (Hutchinson, 2003; Renslo et al., 2006a; Prasad, 2007), including:
- Cyclic isosteres of oxazolidinone, such as isoxazoline and isoxazolinone systems.
- Conformationally restricted versions.
- C5 side-chain-modified analogues.
- Morpholino replacements.
Many of these analogues were accessed using the same, or similar, chemical methodology, with the various parts of linezolid replaced by using a range of different reactants in the synthesis, allowing exploration of the steric, electronic, and hydrogen-bonding requirements for activity (Hutchinson et al., 1993). Other syntheses have been developed from natural precursors, from the so-called ‘chiral pool’,23 such as D-mannitol (Lohray et al., 1999) and L-serine (Einsiedel et al., 2003), or by using alternative chiral synthetic methodology to construct the oxazolidinone ring (Song et al., 2008; Vargas et al., 2009). Some of the newer analogues (patented in 2007) by Pfizer involve even greater synthetic challenges, to match the greater complexity of the oxazolidinone compounds under investigation (Thomas et al., 2007).
4.5.3 Bioavailability
Both linezolid and eperezolid were evaluated for their bioavailability and pharmacokinetic properties in vitro and during pre-clinical trials (Brickner et al., 2008). As was mentioned earlier, the overall profile of linezolid was better than that of eperezolid in these early tests, so it was progressed to the clinic and the vast majority of the clinical pharmacokinetics and pharmacodynamics information in the research literature relates to it (MacGowan, 2003; Vardakas et al., 2009; Dryden, 2011) and its pharmacokinetic properties and clinical outcomes in comparison to similar antibacterial agents, such as vancomycin, daptomycin, and tigecycline (Manfredi and Sabbatani, 2010).
When you consider the structure of linezolid, you can see that it is a neutral molecule with no easily ionised groups and several hydrophobic sections, rendering it suitable for absorption across a biological membrane (both mammalian and bacterial). At the same time, however, it has several atoms with lone pairs of electrons that provide favourable intermolecular interactions with water molecules and so help to increase its aqueous solubility. As a result, linezolid shows excellent oral bioavailability (100%), while its relatively low LogP of 0.55 (Herrmann et al., 2008) enables good aqueous solubility (3.7 mg/mL in phosphate buffer at pH 7) (Brickner et al., 2008), thus offering the possibility of IV administration, especially as the bioavailability of linezolid when administered IV is also 100% (Welshman et al., 2001). This ability to choose between IV and oral administration routes offers the potential for hospitalised patients to return home earlier, once they are able to take an oral dose (Dryden, 2011).
The MIC of linezolid is in the range 0.5–4 mg/L (or μg/mL) for the majority of Gram positive bacteria; when the in vivo concentration achieves four times the MIC, linezolid shows a post-antibiotic effect which, although relatively short, enhances its antibacterial effect (Munckhof et al., 2001). These properties, combined with a half-life (t½) of 3.4–7.4 hours (Dryden, 2011) and time-dependent pharmacokinetics (Zurenko et al., 2001), lead to an oral or IV dose of 600 mg every 12 hours, achieving a steady-state serum concentration (Cmax) of 15–27 mg/L at 0.5–2 hours after administration (Dryden, 2011).
The rate of absorption of linezolid is reduced as a result of oral administration with or directly after food, causing a delay in the achievement of peak plasma concentrations of about 2 hours; co-administration with high-fat-content food also leads to a decrease in the blood concentration. The overall extent of absorption is not affected (with comparable tissue distribution and total bioavailability). Under fasting and non-fasting conditions, such that levels were sufficient to exceed the usual MIC of ≤4 mg/L (Welshman et al., 2001; Islinger et al., 2006), no interaction was observed between antacids and linezolid (Grunder et al., 2006).
Linezolid exhibits around 30% plasma protein binding and a volume of distribution of about 40–50 L, which roughly equates to the total body water content (Dryden, 2011). It exhibits extensive tissue distribution, achieving greater than MIC levels in skin, pulmonary alveolar cells, bone, fat, muscle cells, sweat, saliva, and CSF, with generally similar or higher concentrations in both healthy and inflamed tissues than in plasma (MacGowan, 2003; Vardakas et al., 2009; Dryden, 2011). The importance of this extensive tissue penetration has been linked to the successful treatment of MRSA-related pneumonia, skin and soft tissue infections (SSTIs), Gram positive bacterial orthopedic infections, and even multiresistant Enterococcus faecium-related meningitis (Dryden, 2011). Sufficiently high concentrations of linezolid are reached in the aqueous humour and vitreous of the eye after oral administration to achieve antibacterial action against strains of staphylococci responsible for ocular infections (Horcajada et al., 2009).
The absorption of linezolid in children appears to be similar to that in adults, whereas the distribution and plasma clearance are enhanced in infants and children (MacGowan, 2003). A pharmacokinetic model was established which showed that both the elderly and patients with lower body weights have altered distribution and clearance (Abe et al., 2009). A higher plasma concentration is achieved in adult females than in males, alongside a lower total clearance and volume of distribution, whereas the serum half-life is the same in both (MacGowan, 2003; Dryden, 2011).
Linezolid is metabolised, by non-enzymatic oxidation of the morpholine ring (Scheme 4.5.2), into two major inactive metabolites, both of which are excreted in the urine, along with around 30% linezolid, accounting for >80% of the dose (MacGowan, 2003; Vardakas et al., 2009).
Scheme 4.5.2 Non-enzymatic metabolism of linezolid, resulting in two major metabolites (MacGowan, 2003)
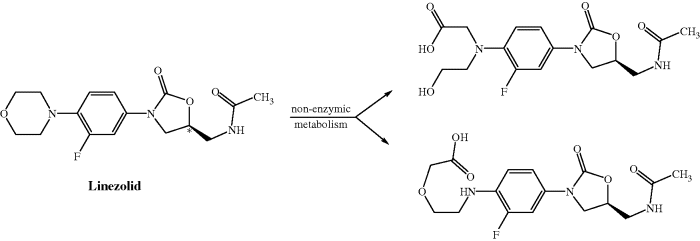
The faecal excretion route is non-existent for linezolid, and very minor for the metabolites, and cytochrome P450 (CYP) enzymes do not appear to be involved in linezolid metabolism, so inducement or inhibition of liver enzymes is not expected, and there is an associated low risk of interactions with the usual CYP-dependent suspects, such as cimetidine and phenytoin (Vardakas et al., 2009).
Linezolid is generally accepted to be bacteriostatic, but it shows clinical bactericidal activity against most strains of streptococci and some other bacteria (Zurenko et al., 2001), perhaps explained by a linezolid-induced decrease in the amount of virulence factor released, which increases bacterial susceptibility to neutrophil phagocytosis (Gemmell and Ford, 2002; Bernardo et al., 2004).
4.5.4 Mode of Action and Selectivity
Linezolid acts by preventing protein synthesis, but in a different manner to the agents we have studied previously in this section. Although it competes with chloramphenicol in binding assays with the 50S ribosomal subunit, it does not have the same mode of action, as it does not inhibit binding to the peptidyl transferase A site (described in Subsection 4.4.4). It has taken a number of years to identify the most likely binding site and the mode of action of linezolid and related oxazolidinones (Leach et al., 2011).
So where does it bind and what is its mode of action? By studying the 50S ribosomal subunit of representative bacterial ribosomes (Ippolito et al., 2008; Wilson et al., 2008), X-ray crystallography has shown that linezolid binds to the A site, close to the catalytic centre, and disturbs the binding of the charged tRNA to this site. More specifically, linezolid binds in a pocket formed by eight RNA residues that are conserved across all bacterial 50S ribosomes: U2585, A2503, U2504, G2505, U2506, G2061, A2451, and C2452 (using the numbering system of Escherichia coli) (Wilson et al., 2008). Although most of these residues do not need to move to accommodate linezolid, there is an induced-fit element to linezolid binding, with certain residues moving in order to better interact with linezolid. This induced movement of residues results in alternate orientations of these bases and could be responsible for a decreased ability to bind the next charged tRNA to the A site, or linezolid binding partly in the A site may prohibit tRNA binding (Ippolito et al., 2008; Wilson et al., 2008).
Selectivity for a bacterial target over a mammalian one is crucial to the clinical success of an antibacterial agent. The examples of bacterial protein synthesis inhibitors we have already studied in this section show selectivity due to a variety of reasons, including:
- Poor uptake into mammalian cells compared to bacterial cells.
- The incorporation of specific functional groups for optimum selectivity.
- Poor binding to the mammalian ribosome.
As we have seen, the aminoglycosides, macrolides, tetracyclines, and chloramphenicol are all antibiotics: they are natural molecules from a microbial source which have benefitted from Nature’s design expertise to optimise the structures for their antibacterial effect. In comparison, linezolid is an antibacterial agent that is imperfect, being of human design (with little knowledge of the target or mode of action for guidance), and this is reflected in its lack of absolute selectivity for bacterial over mammalian protein synthesis. Linezolid has no effect upon mammalian cytoplasmic protein synthesis, but inhibits mitochondrial protein synthesis (Leach et al., 2007), which is linked to the serious adverse effect of myelosuppression when used for an extended period (De Vriese et al., 2006), causing concerns and reviews of its safety (Vinh and Rubenstein, 2009). In previous subsections, we saw how the same issue (the inhibition of mitochondrial protein synthesis) restricts the use of chloramphenicol, while some of the adverse effects of tetracyclines are due to their actions upon mitochondrial processes, and aminoglycosides also exhibit toxicity – despite Nature’s expertise, perhaps it doesn’t always get it right!
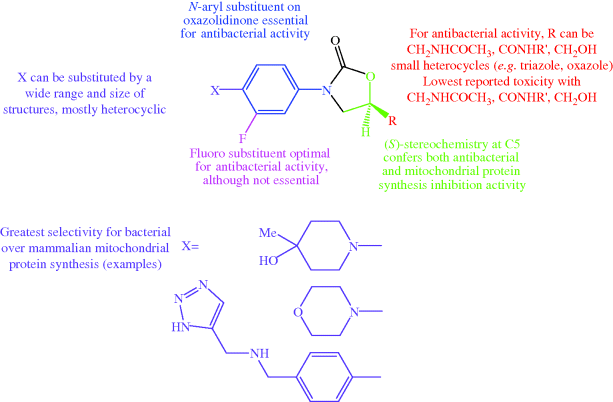
The huge number of linezolid analogues that have been synthesised and studied (Poce et al., 2008) have fine-tuned the structural features and lipophilicity, while reducing the undesirable effects due to inhibition of human mitochondrial protein synthesis and monoamine oxidase A, enabling an updated overall structure–activity relationship (Katritzky et al., 2004; Renslo et al., 2006b; Prasad, 2007) and toxicity–structure relationship (Renslo, 2010) to be developed (Figure 4.5.4).
Figure 4.5.4 Combined structure–activity and structure–toxicity relationships developed after extensive study of linezolid analogues (Renslo et al., 2006b; Prasad, 2007; Renslo, 2010)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


