INTRODUCTION
LEARNING OBJECTIVES
After studying this chapter you should be able to:
Understand the physiologic consequences of platelet activation.
Name the zymogens and cofactors in the coagulation cascade and know how they relate to one another.
Identify the mechanisms that limit blood clot formation.
Describe the laboratory tests that are most useful in the diagnosis and monitoring of bleeding disorders.
Understand the mechanisms underlying the action of anticoagulant drugs.
Hemostasis is a vital host defense system that has evolved in man and other vertebrates to maintain the integrity of a high-pressure closed circulatory system. Following a break in the vasculature, the formation of a blood clot either prevents hemorrhage or lessens its extent. An exquisitely modulated orchestration of cells and plasma proteins enables clot formation to be rapid, self-limited, and reversible. The classic paradigm of blood coagulation begins with the formation of a platelet thrombus at the site of vessel injury. Initially, platelets adhere to collagen exposed on the denuded surface of the injured vessel. This adhesion event then triggers activation of platelets by collagen, thrombin generated by tissue factor, or both, allowing them to aggregate and form a tight platelet plug that stanches the further egress of blood from the injured vessel. The clot is then reinforced and strengthened by the activation of the coagulation cascade, which results in the deposition of insoluble polymers composed of fibrin. The recruitment of specific inhibitors prevents unwanted extension of the clot beyond the site of injury. The final step, fibrinolysis, enables the vessel to be recanalized, thereby restoring blood flow. Recent studies have shown that these phases are less clearly demarcated in vivo than this idealized scenario would suggest. Indeed, the use of specific fluorescent molecular markers clearly shows that fibrin deposition occurs as the platelet plug is being formed.
Hemostatic system
latent but poised for immediate participation
Blood coagulation
limited to region of vessel injury
This chapter will first present the dynamic sequence of events involved in formation of the platelet thrombus and the initiation, propagation, limitation, and lysis of the fibrin clot. This conceptual framework will guide the discussion of the pathophysiology, diagnosis, and treatment of bleeding and thrombotic disorders in this and the following chapters.
PLATELET ADHESION AND ACTIVATION
Plasma contains a large, elongated protein called von Willebrand factor (vWF) that forms multimeric polymers with molecular weights as high as 15 million Daltons. As discussed later in this chapter, vWF binds to and transports factor VIII, the protein deficient in patients with classic hemophilia. In addition, vWF fulfills a vital role in the formation of the primary platelet plug. It has binding sites for collagen as well as for a glycoprotein complex on the surface of platelets, GPIb-IX-V (hereafter referred to simply as the GPIb complex). Under high shear stresses, such as occur within flowing blood, vWF “unwinds” to an extended conformation in which it can bind to both platelet GPIb complexes and to collagen exposed by damage to the blood vessel. Thus, vWF is a kind of molecular “glue” that allows platelets to adhere to injured vessel walls.
The platelet is activated by adhesion as well as by thrombin generated by tissue factor. Thrombin activates platelets by cleaving the platelet thrombin “receptor” in its extracellular domain, causing a conformational change that transduces an activating signal. As depicted in Figure 13-1A, activation entails a sudden and irreversible change in platelet shape from a smooth disc to a spiky “sea urchin” with multiple pseudopods, markedly increasing the surface area of the platelet. Activation also leads to a conformational change in another platelet membrane glycoprotein, GPIIb/IIIa, which allows it to bind fibrinogen. Fibrinogen is bivalent and can therefore form bridges between GPIIb/IIIa receptors on two different platelets. This cross-linking event is the crucial step in platelet aggregation and the propagation of the primary platelet plug.
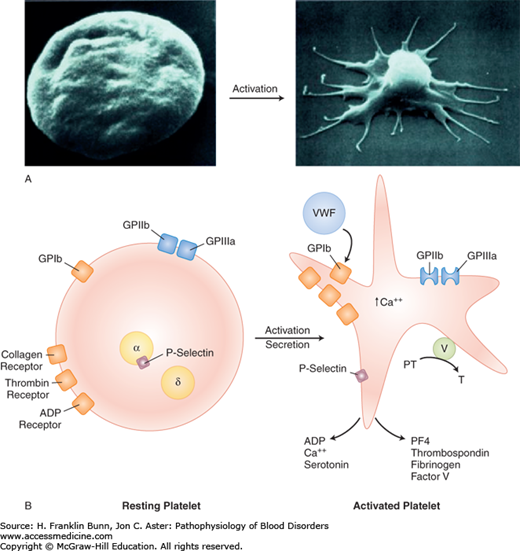
In addition to adhesion and thrombin, platelet activation can be triggered by other physiologically relevant stimuli, such as adenosine diphosphate (ADP). ADP is one of a number of molecules released from the dense granules of activated platelets. In the local milieu of the platelet plug, ADP binds to receptors on nearby unstimulated platelets and causes their activation. This is one of several amplification loops that ramp-up the hemostatic process in the immediate vicinity of a vascular injury, enabling rapid clot formation.
Activation of platelets also results in the release of arachidonic acid, a long-chain polyunsaturated fatty acid, from phospholipids on the plasma membrane. Arachidonic acid is converted to thromboxane A2, a potent inducer of platelet aggregation, by a series of enzymes including cyclooxygenase. Inhibition of platelet cyclooxygenase explains the ability of aspirin to suppress platelet activation.
When platelets are activated, another morphologically distinct organelle, the alpha granule, releases proteins that contribute to hemostasis, including platelet factor 4, thrombospondin, fibrinogen, and factor V. In addition, P-selectin in the alpha granule membrane is translocated to the surface plasma membrane of the activated platelet where it plays an important role in the interaction of platelets with other cells, including leukocytes and the vascular endothelium. These processes provide an important link to the soluble coagulation system. The factor Xa receptor, defined by phospholipid on the surface of the activated platelet, cooperates with factor V in the generation of thrombin, as shown in Figure 13-1B and discussed in more detail in the following section.
THE BLOOD COAGULATION CASCADE
Just as the formation of the platelet plug is a highly cooperative process, so is the concomitant formation of the fibrin clot. Amplification of a small signal (vascular injury) is transduced by a series of molecular interactions into a fibrin clot. The protagonists in this dramatic process are a set of plasma proteins that normally circulate as inactive zymogens. Figure 13-2 depicts a prototypical or consensus pathway. When the coagulation cascade is activated, either in vivo or in the test tube, a zymogen is converted into an active proteolytic enzyme capable of specifically cleaving the next zymogen in the set, and so on. This highly controlled limited proteolysis leads to the eventual formation of thrombin and generation of the fibrin clot.
During the last century, biochemical studies of in vitro clot formation have gradually led to the identification of the specific proteins that participate in the coagulation cascade, along with in-depth understanding of how they interact in a precisely controlled and orchestrated fashion. A substantial portion of this cumulative knowledge has come from rigorous investigation of families with inherited defects of specific coagulation proteins (see Chapter 15).
Figure 13-3 depicts the coagulation proteins involved in clot formation prior to the formation of fibrin. The zymogens, shown in image A (prothrombin and factors VII, IX, X, and XI), are synthesized in the liver. As this figure demonstrates, they have some striking structural similarities. All have a signal peptide at the N-terminus, and except for factor XI, a propeptide is immediately adjacent. These N-terminal portions are cleaved within the hepatocyte prior to the release of the mature zymogen into the circulation. All five zymogens have a large catalytic domain at the C-terminus that, when activated, functions as a serine protease that specifically cleaves and activates the next protein in the coagulation cascade. Four of these zymogens, factors VII, IX, X, and prothrombin, undergo a crucial post-translational modification in the Golgi body prior to secretion into the plasma. Specific glutamic acid residues in the region adjacent to the propeptide undergo vitamin K–dependent carboxylation. This additional negative charge facilitates the binding of calcium ions, an essential cofactor for optimal function.
FIGURE 13-3
Proteins in the proximal coagulation cascade. A) Zymogens are converted to active enzymes by proteolytic cleavage. Blue rectangles at left = signal peptides; • • • • • = propeptide; Y = specific sites of glutamic acid carboxylation. The curved arrows to the right show sites of cleavage releasing the C-terminal catalytic domain (long blue rectangle on right), which now functions as an active serine protease. B) Cofactors serve as docking sites for zymogens. Tissue factor is a transmembrane protein. (The lipid bilayer is shown as a pair of vertical rectangles.) Factors VIII and V are activated by proteolytic cleavage at sites shown by the curved arrows. (Modified with permission from Furie B and Furie BC. The molecular basis of blood coagulation. Cell. 1988. 53: 505-518.)
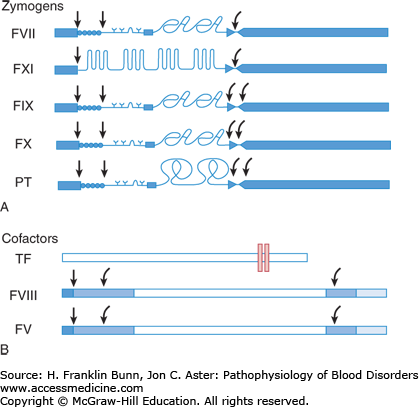
Protein cofactors play an equally important role in the regulation of the coagulation cascade. They bind to platelet and endothelial cell membranes, serving as docking sites for the zymogens and contributing importantly to the amplification that is essential for the rapid formation of the fibrin clot. As shown in Figure 13-3B, tissue factor is a transmembrane protein on the surface of extravascular cells, cells within the vessel wall, and circulating microparticles. Like the zymogens mentioned earlier, factors VIII and V are actually pro-cofactors—soluble plasma proteins that require proteolytic cleavage for activation.
In the test tube, the formation of the fibrin clot proceeds by the intrinsic pathway depicted in Figure 13-4
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


