OBJECTIVES
After studying this chapter, you should be able to:
Name the prominent cellular organelles and state their functions in cells.
Name the building blocks of the cellular cytoskeleton and state their contributions to cell structure and function.
Name the intercellular and cellular to extracellular connections.
Define the processes of exocytosis and endocytosis, and describe the contribution of each to normal cell function.
Define proteins that contribute to membrane permeability and transport.
Recognize various forms of intercellular communication and describe ways in which chemical messengers (including second messengers) affect cellularphysiology.
INTRODUCTION
The cell is the fundamental working unit of all organisms. In humans, cells can be highly specialized in both structure and function; alternatively, cells from different organs can share features and function. In the previous chapter, some basic principles of biophysics and the catabolism and metabolism of building blocks found in the cell were examined. In some of those discussions, how the building blocks could contribute to basic cellular physiology (eg, DNA replication, transcription, and translation) were discussed. In this chapter, more of the fundamental aspects of cellular and molecular physiology will be reviewed. Additional aspects that concern specialization of cellular and molecular physiology are considered in the next chapters concerning immune function and excitable cells and within the sections that highlight each physiologic system.
FUNCTIONAL MORPHOLOGY OF THE CELL
A basic knowledge of cell biology is essential to an understanding of the organ systems and the way they function in the body. A key tool for examining cellular constituents is the microscope. A light microscope can resolve structures as close as 0.2 μm, while an electron microscope can resolve structures as close as 0.002 μm. Although cell dimensions are quite variable, this resolution can provide a good look at the inner workings of the cell. The advent of common access to phase contrast, fluorescent, confocal, and many other microscopy techniques along with specialized probes for both static and dynamic cellular structures further expanded the examination of cell structure and function. Equally revolutionary advances in modern biophysical, biochemical, and molecular biological techniques have also greatly contributed to our knowledge of the cell.
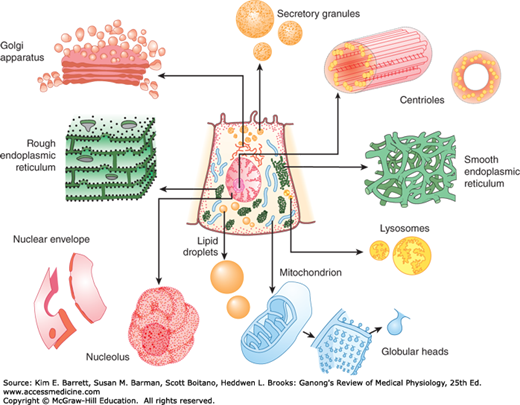
The specialization of the cells in the various organs is considerable, and no cell can be called “typical” of all cells in the body. However, a number of structures (organelles) are common to most cells. These structures are shown in Figure 2–1. Many of them can be isolated by ultracentrifugation combined with other techniques. When cells are homogenized and the resulting suspension is centrifuged, the nuclei sediment first, followed by the mitochondria. High-speed centrifugation that generates forces of 100,000 times gravity or more causes a fraction made up of granules called the microsomes to sediment. This fraction includes organelles such as the ribosomes and peroxisomes.
FIGURE 2–1
Cross-sectional diagram of a hypothetical cell as seen with the light microscope. Individual organelles are expanded for closer examination. (Adapted with permission from Bloom and Fawcett. Reproduced with permission from Junqueira LC, Carneiro J, Kelley RO: Basic Histology, 9th ed. New York, NY: McGraw-Hill; 1998.)
The membrane that surrounds the cell is a remarkable structure. It is made up of lipids and proteins and is semipermeable, allowing some substances to pass through it and excluding others. However, its permeability can also be varied because it contains numerous regulated ion channels and other transport proteins that can change the amounts of substances moving across it. It is generally referred to as the plasma membrane. The nucleus and other organelles in the cell are bound by similar membranous structures.
Although the chemical structures of membranes and their properties vary considerably from one location to another, they have certain common features. They are generally about 7.5 nm (75 angstroms [Å]) thick. The major lipids are phospholipids such as phosphatidylcholine, phosphatidylserine, and phosphatidylethanolamine. The shape of the phospholipid molecule reflects its solubility properties: the “head” end of the molecule contains the phosphate portion and is relatively soluble in water (polar, hydrophilic) and the “tail” ends are relatively insoluble (nonpolar, hydrophobic). The possession of both hydrophilic and hydrophobic properties makes the lipid an amphipathic molecule. In the membrane, the hydrophilic ends of the molecules are exposed to the aqueous environment that bathes the exterior of the cells and the aqueous cytoplasm; the hydrophobic ends meet in the water-poor interior of the membrane (Figure 2–2). In prokaryotes (ie, bacteria in which there is no nucleus), the membranes are relatively simple, but in eukaryotes (cells containing nuclei), cell membranes contain various glycosphingolipids, sphingomyelin, and cholesterol in addition to phospholipids and phosphatidylcholine.
FIGURE 2–2
Organization of the phospholipid bilayer and associated proteins in a biologic membrane. The phospholipid molecules that make up the membrane each have two hydrophobic fatty acid chains attached to a hydrophilic phosphate head. Individual proteins take on different shapes and positions in the cell. Many are integral proteins, extending into the membrane or peripheral proteins that are attached to the inside or outside (not shown) of the membrane. Proteins can be modified (eg, with carbohydrate chains). Many specific protein attachments and cholesterol that are commonly found in the bilayer are omitted for clarity.
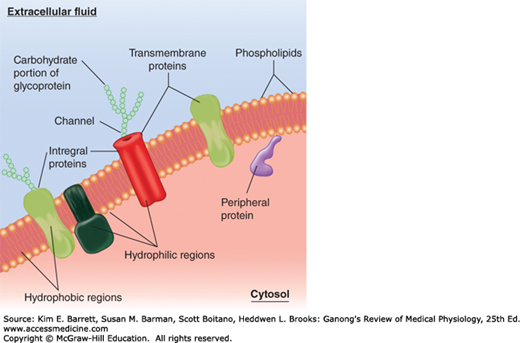
Many different proteins are embedded in the membrane. They exist as separate globular units and many pass through or are embedded in one leaflet of the membrane (integral proteins), whereas others (peripheral proteins) are associated with the inside or outside of the membrane (Figure 2–2). The amount of protein varies significantly with the function of the cell but makes up on average 50% of the mass of the membrane; that is, there is about one protein molecule per 50 of the much smaller phospholipid molecules. The proteins in the membrane carry out many functions. Some are cell adhesion molecules (CAMs) that anchor cells to their neighbors or to basal laminas. Some proteins function as pumps, actively transporting ions across the membrane. Other proteins function as carriers, transporting substances down electrochemical gradients by facilitated diffusion. Still others are ion channels, which, when activated, permit the passage of ions into or out of the cell. The role of the pumps, carriers, and ion channels in transport across the cell membrane is discussed below. Proteins in another group function as receptors that bind ligands or messenger molecules, initiating physiologic changes inside the cell. Proteins also function as enzymes, catalyzing reactions at the surfaces of the membrane. Examples from each of these groups are discussed later in this chapter.
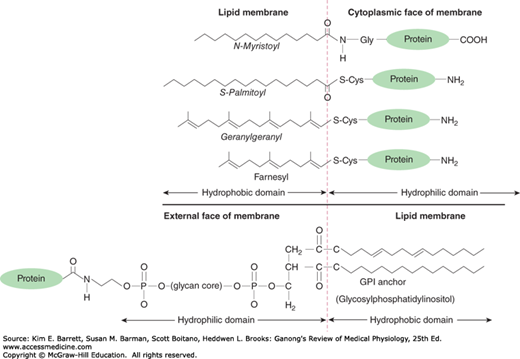
The hydrophobic portions of the proteins are usually located in the interior of the membrane, whereas the charged, hydrophilic portions are located on the surfaces. Peripheral proteins are attached to the surfaces of the membrane in various ways. One common way is attachment to glycosylated forms of phosphatidylinositol. Proteins held by these glycosylphosphatidylinositol (GPI) anchors (Figure 2–3) include enzymes such as alkaline phosphatase, various antigens, a number of CAMs, and proteins that combat cell lysis by complement. Over 250 GPI-linked cell surface proteins have now been described in humans. Other proteins are lipidated, that is, they have specific lipids attached to them (Figure 2–3). Proteins may be myristoylated, palmitoylated, or prenylated (ie, attached to geranylgeranyl or farnesyl groups).
FIGURE 2–3
Protein linkages to membrane lipids. A variety of lipid modifications can occur at amino or carboxy terminals of proteins attached to cytosolic side of the plasma membrane. Many proteins associated with the external side of the plasma membrane can be attached via glycosylated forms of phosphatidylinositol (eg, GPI anchors).
The protein structure—and particularly the enzyme content—of biologic membranes varies not only from cell to cell, but also within the same cell. For example, some of the enzymes embedded in cell membranes are different from those in mitochondrial membranes. In epithelial cells, the enzymes in the cell membrane on the mucosal surface differ from those in the cell membrane on the basal and lateral margins of the cells; that is, the cells are polarized, Such polarization makes directional transport across epithelia possible. The membranes are dynamic structures, and their constituents are being constantly renewed at different rates. Some proteins are anchored to the cytoskeleton, but others move laterally in the membrane.
Underlying most cells is a thin, “fuzzy” layer plus some fibrils that collectively make up the basement membrane or, more properly, the basal lamina. The basal lamina and, more generally, the extracellular matrix are made up of many proteins that hold cells together, regulate their development, and determine their growth. These include collagens, laminins, fibronectin, tenascin, and various proteoglycans.
Over a billion years ago, aerobic bacteria were engulfed by eukaryotic cells and evolved into mitochondria, providing the eukaryotic cells with the ability to form the energy-rich compound ATP by oxidative phosphorylation. Mitochondria perform other functions, including a role in the regulation of apoptosis (programmed cell death), but oxidative phosphorylation is the most crucial. Each eukaryotic cell can have hundreds to thousands of mitochondria. In mammals, they are generally depicted as sausage-shaped organelles (Figure 2–1), but their shape can be quite dynamic. Each has an outer membrane, an intermembrane space, an inner membrane, which is folded to form shelves (cristae), and a central matrix space. The enzyme complexes responsible for oxidative phosphorylation are lined up on the cristae (Figure 2–4).
FIGURE 2–4
Components involved in oxidative phosphorylation in mitochondria and their origins. As enzyme complexes I through IV convert 2-carbon metabolic fragments to CO2 and H2O, protons (H+) are pumped into the intermembrane space. The protons diffuse back to the matrix space via complex V, ATP synthase (AS), in which ADP is converted to ATP. The enzyme complexes are made up of subunits coded by mitochondrial DNA (mDNA) and nuclear DNA (nDNA), and the figures document the contribution of each DNA to the complexes.
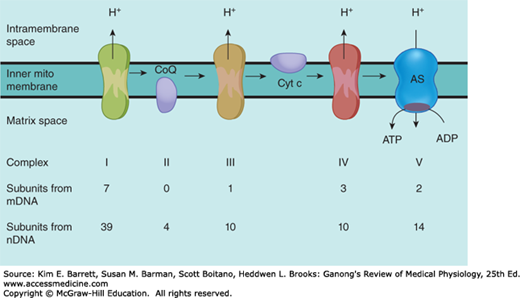
Consistent with their origin from aerobic bacteria, the mitochondria have their own genome. There is much less DNA in the mitochondrial genome than in the nuclear genome, and 99% of the proteins in the mitochondria are the products of nuclear genes, but mitochondrial DNA is responsible for certain key components of the pathway for oxidative phosphorylation. Specifically, human mitochondrial DNA is a double-stranded circular molecule containing approximately 16,500 base pairs (compared with over a billion in nuclear DNA). It codes for 13 protein subunits that are associated with proteins encoded by nuclear genes to form four enzyme complexes plus two ribosomal and 22 transfer RNAs that are needed for protein production by the intramitochondrial ribosomes.
The enzyme complexes responsible for oxidative phosphorylation illustrate the interactions between the products of the mitochondrial genome and the nuclear genome. For example, complex I, reduced nicotinamide adenine dinucleotide dehydrogenase (NADH), is made up of seven protein subunits coded by mitochondrial DNA and 39 subunits coded by nuclear DNA. The origin of the subunits in the other complexes is shown in Figure 2–4. Complex II, succinate dehydrogenase-ubiquinone oxidoreductase; complex III, ubiquinonecytochrome c oxidoreductase; and complex IV, cytochrome c oxidase, act with complex I, coenzyme Q, and cytochrome c to convert metabolites to CO2 and water. Complexes I, III, and IV pump protons (H+) into the intermembrane space during this electron transfer. The protons then flow down their electrochemical gradient through complex V, ATP synthase, which harnesses this energy to generate ATP.
As zygote mitochondria are derived from the ovum, their inheritance is maternal. This maternal inheritance has been used as a tool to track evolutionary descent. Mitochondria have an ineffective DNA repair system, and the mutation rate for mitochondrial DNA is over 10 times the rate for nuclear DNA. A large number of relatively rare diseases have been traced to mutations in mitochondrial DNA. These include disorders of tissues with high metabolic rates in which energy production is defective as a result of abnormalities in the production of ATP, as well as other disorders (Clinical Box 2–1).
CLINICAL BOX 2–1 Mitochondrial Diseases
Mitochondrial diseases encompass at least 40 diverse disorders that are grouped because of their links to mitochondrial failure. These diseases can occur following inheritance or spontaneous mutations in mitochondrial or nuclear DNA that lead to altered functions of the mitochondrial proteins (or RNA). Depending on the target cell and/or tissues affected, symptoms resulting from mitochondrial diseases may include altered motor control; altered muscle output; gastrointestinal dysfunction; altered growth; diabetes; seizures; visual/hearing problems; lactic acidosis; developmental delays; and susceptibility to infection or cardiac, liver, and respiratory disease. Although there is evidence for tissue-specific isoforms of mitochondrial proteins, mutations in these proteins do not fully explain the highly variable patterns or targeted organ systems observed with mitochondrial diseases.
THERAPEUTIC HIGHLIGHTSWith the diversity of disease types and the overall importance of mitochondria in energy production, it is not surprising that there is no single cure for mitochondrial diseases and focus remains on treating the symptoms when possible. For example, in some mitochondrial myopathies (ie, mitochondrial diseases associated with neuromuscular function), physical therapy may help extend the range of movement of muscles and improve dexterity.
In the cytoplasm of the cell there are large, somewhat irregular structures surrounded by membranes. The interior of these structures, which are called lysosomes, is more acidic than the rest of the cytoplasm, and external material such as endocytosed bacteria, as well as worn-out cell components, are digested in them. The interior is kept acidic by the action of a proton pump, or H+ ATPase. This integral membrane protein uses the energy of ATP to move protons from the cytosol up their electrochemical gradient and keep the lysosome relatively acidic, near pH 5.0. Lysosomes can contain over 40 types of hydrolytic enzymes, some of which are listed in Table 2–1. Not surprisingly, these enzymes are all acid hydrolases, in that they function best at the acidic pH of the lysosomal compartment. This can be a safety feature for the cell; if the lysosomes were to break open and release their contents, the enzymes would not be efficient at the near neutral cytosolic pH 7.2, and thus would be unable to digest cytosolic targets they may encounter. Diseases associated with lysosomal dysfunction are discussed in Clinical Box 2–2.
| Enzyme | Substrate |
|---|---|
| Ribonuclease | RNA |
| Deoxyribonuclease | DNA |
Phosphatase Glycosidases Arylsulfatases | Phosphate esters Complex carbohydrates: glycosides and polysaccharides Sulfate esters |
| Collagenase | Collagens |
| Cathepsins | Proteins |
CLINICAL BOX 2–2 Lysosomal Diseases
When a lysosomal enzyme is congenitally absent, the lysosomes become engorged with the material the enzyme normally degrades. This eventually leads to one of the lysosomal diseases (also called lysosomal storage diseases). There are over 50 such diseases currently recognized. For example, Fabry disease is caused by a deficiency in α-galactosidase; Gaucher disease is caused by a deficiency in β-galactocerebrosidase; and Tay-Sachs disease, which causes mental retardation and blindness, is caused by the loss of hexosaminidase A, a lysosomal enzyme that catalyzes the biodegradation of gangliosides (fatty acid derivatives). Such individual lysosomal diseases are rare, but they are serious and can be fatal.
THERAPEUTIC HIGHLIGHTSSince there are many different lysosomal disorders, treatments vary considerably and “cures” remain elusive for most of these diseases. Much of the care is focused on managing symptoms of each specific disorder. Enzyme replacement therapy has shown to be effective for certain lysosomal diseases, including Gaucher disease and Fabry disease. However, the long-term effectiveness and the tissue-specific effects of many of the enzyme replacement treatments have not yet been established. Recent alternative approaches include bone marrow or stem cell transplantation. Again, medical advances are still necessary to fully combat this group of diseases.
Peroxisomes are 0.5 μm in diameter, are surrounded by a membrane, and contain enzymes that can either produce H2O2 (oxidases) or break it down (catalases). Proteins are directed to the peroxisome by a unique signal sequence with the help of protein chaperones, peroxins. The peroxisome membrane contains a number of peroxisome-specific proteins that are concerned with transport of substances into and out of the matrix of the peroxisome. The matrix contains more than 40 enzymes, which operate in concert with enzymes outside the peroxisome to catalyze a variety of anabolic and catabolic reactions (eg, breakdown of lipids). Peroxisomes can form by budding of the endoplasmic reticulum, or by division. A number of synthetic compounds were found to cause proliferation of peroxisomes by acting on receptors in the nuclei of cells. These peroxisome proliferator-activated receptors (PPARs) are members of the nuclear receptor superfamily. When activated, they bind to DNA, producing changes in the production of mRNAs. The known effects for PPARs are extensive and can affect most tissues and organs.
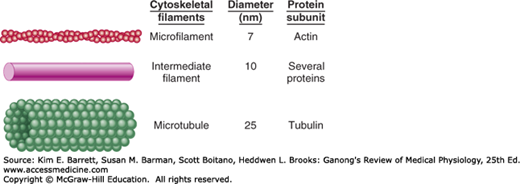
All cells have a cytoskeleton, a system of fibers that not only maintains the structure of the cell but also permits it to change shape and move. The cytoskeleton is made up primarily of microtubules, intermediate filaments, and microfilaments (Figure 2–5), along with proteins that anchor them and tie them together. In addition, proteins and organelles move along microtubules and microfilaments from one part of the cell to another, propelled by molecular motors.
FIGURE 2–5
Cytoskeletal elements of the cell. Artistic impressions that depict the major cytoskeletal elements are shown on the left, with approximate diameters and protein subunits of these elements are listed for comparison. (Reproduced with permission from Widmaier EP, Raff H, Strang KT: Vander’s Human Physiology: The Mechanisms of Body Function, 11th ed. New York, NY: McGraw-Hill; 2008.)
Microtubules (Figure 2–5 and Figure 2–6) are long, hollow structures with 5 nm walls surrounding a cavity 15 nm in diameter. They are made up of two globular protein subunits: α- and β-tubulin. A third subunit, γ-tubulin, is associated with the production of microtubules by the centrosomes. The α and β subunits form heterodimers, which aggregate to form long tubes made up of stacked rings, with each ring usually containing 13 subunits. The tubules interact with guanosine triphosphate (GTP) to facilitate their formation. Although microtubule subunits can be added to either end, microtubules are polar with assembly predominating at the “+” end and disassembly predominating at the “–” end. Both processes occur simultaneously in vitro. The growth of microtubules is temperature sensitive (disassembly is favored under cold conditions) as well as under the control of a variety of cellular factors that can directly interact with microtubules in the cell.
FIGURE 2–6
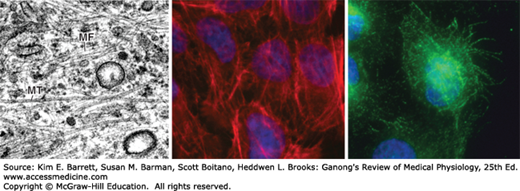
Microfilaments and microtubules. Electron micrograph (Left) of the cytoplasm of a fibroblast, displaying actin microfilaments (MF) and microtubules (MT). Fluorescent micrographs of airway epithelial cells displaying actin microfilaments stained with phalloidin (Middle) and microtubules visualized with an antibody to β-tubulin (Right). Both fluorescent micrographs are counterstained with Hoechst dye (blue) to visualize nuclei. Note the distinct differences in cytoskeletal structure. (Electron 70 micrograph used with permission of E Katchburian; fluorescent micrographs used with permission of Stephanie Manberg.)
Because of their constant assembly and disassembly, microtubules are a dynamic portion of the cytoskeleton. They provide the tracks along which several different molecular motors move transport vesicles, organelles such as secretory granules, and mitochondria from one part of the cell to another. They also form the spindle, which moves the chromosomes in mitosis. Cargo can be transported in either direction on microtubules.
There are several drugs available that disrupt cellular function through interaction with microtubules. Microtubule assembly is prevented by colchicine and vinblastine. The anticancer drug paclitaxel (Taxol) binds to microtubules and makes them so stable that organelles cannot move. Mitotic spindles cannot form, and the cells die.
Intermediate filaments (Figure 2–5) are 8–14 nm in diameter and are made up of various subunits. Some of these filaments connect the nuclear membrane to the cell membrane. They form a flexible scaffolding for the cell and help it resist external pressure. In their absence, cells rupture more easily, and when they are abnormal in humans, blistering of the skin is common. The proteins that make up intermediate filaments are cell-type specific, and are thus frequently used as cellular markers. For example, vimentin is a major intermediate filament in fibroblasts, whereas cytokeratin is expressed in epithelial cells.
Microfilaments (Figures 2–5 and 2–6) are long solid fibers with a 5–9 nm diameter that are made up of actin. Although actin is most often associated with muscle contraction, it is present in all types of cells. It is the most abundant protein in mammalian cells, sometimes accounting for as much as 15% of the total protein in the cell. Its structure is highly conserved; for example, 88% of the amino acid sequences in yeast and rabbit actin are identical. Actin filaments polymerize and depolymerize in vivo, and it is not uncommon to find polymerization occurring at one end of the filament while depolymerization is occurring at the other end. Filamentous (F) actin refers to intact microfilaments and globular (G) actin refers to the unpolymerized protein actin subunits. F-actin fibers attach to various parts of the cytoskeleton and can interact directly or indirectly with membrane-bound proteins. They reach to the tips of the microvilli on the epithelial cells of the intestinal mucosa. They are also abundant in the lamellipodia that cells put out when they crawl along surfaces. The actin filaments interact with integrin receptors and form focal adhesion complexes, which serve as points of traction with the surface over which the cell pulls itself. In addition, some molecular motors use microfilaments as tracks.
The molecular motors that move proteins, organelles, and other cell parts (collectively referred to as “cargo”) to all parts of the cell are 100–500 kDa ATPases. They attach to their cargo at one end of the molecule and to microtubules or actin polymers with the other end, sometimes referred to as the “head.” They convert the energy of ATP into movement along the cytoskeleton, taking their cargo with them. There are three super families of molecular motors: kinesin, dynein, and myosin. Examples of individual proteins from each superfamily are shown in Figure 2–7. It is important to note that there is extensive variation among superfamily members, allowing for the specialization of function (eg, choice of cargo, cytoskeletal filament type, and/or direction of movement).
FIGURE 2–7
Examples of molecular motors. Conventional kinesin is shown attached to cargo, in this case a membrane-bound organelle (light blue). Cytoplasmic dynein is shown in isolation. Myosin V and its ability to “walk” along a microfilament are displayed in a two-part sequence. Note that the “heads” of each of the motors hydrolyze ATP and use the energy to produce motion.
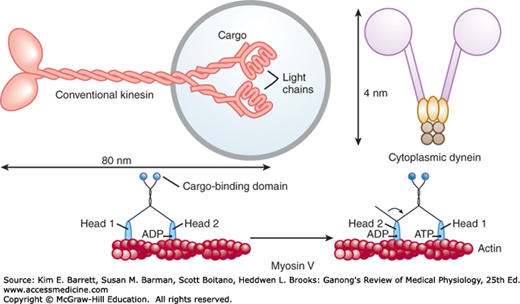
The conventional form of kinesin is a double headed molecule that tends to move its cargo toward the “+” ends of microtubules. One head binds to the microtubule and then bends its neck while the other head swings forward and binds, producing almost continuous movement. Some kinesins are associated with mitosis and meiosis. Other kinesins perform different functions, including, in some instances, moving cargo to the “–” end of microtubules. Dyneins have two heads, with their neck pieces embedded in a complex of proteins. Cytoplasmic dyneins have a function like that of conventional kinesin, except they tend to move particles and membranes to the “–” end of the microtubules. The multiple forms of myosin in the body are divided into 18 classes. The heads of myosin molecules bind to actin and produce motion by bending their neck regions (myosin II) or walking along microfilaments, one head after the other (myosin V). In these ways, they perform functions as diverse as contraction of muscle and cell migration.
Near the nucleus in the cytoplasm of eukaryotic animal cells is a centrosome. The centrosome is made up of two centrioles and surrounding amorphous pericentriolar material. The centrioles are short cylinders arranged so that they are at right angles to each other. Microtubules in groups of three run longitudinally in the walls of each centriole (Figure 2–1). Nine of these triplets are spaced at regular intervals around the circumference.
The centrosomes are microtubule-organizing centers (MTOCs) that contain γ-tubulin. The microtubules grow out of this γ-tubulin in the pericentriolar material. When a cell divides, the centrosomes duplicate themselves, and the pairs move apart to the poles of the mitotic spindle, where they monitor the steps in cell division. In multinucleate cells, a centrosome is near each nucleus.
Cilia are specialized cellular projections that are used by unicellular organisms to propel themselves through liquid and by multicellular organisms to propel mucus and other substances over the surface of various epithelia. Additionally, virtually all cells in the human body contain a primary cilium that emanates from the surface. The primary cilium serves as a sensory organelle that receives both mechanical and chemical signals from other cells and the environment. Cilia are functionally indistinct from the eukaryotic flagella of sperm cells. Within the cilium there is an axoneme that comprises a unique arrangement of nine outer microtubule doublets and two inner microtubules (“9+2” arrangement). Along this cytoskeleton is axonemal dynein. Coordinated dynein–microtubule interactions within the axoneme are the basis of ciliary and sperm movement. At the base of the axoneme and just inside lies the basal body. It has nine circumferential triplet microtubules, like a centriole, and there is evidence that basal bodies and centrioles are interconvertible. A wide variety of diseases and disorders arise from dysfunctional cilia (Clinical Box 2–3).
Cells are attached to the basal lamina and to each other by CAMs that are prominent parts of the intercellular connections described below. The unique structural and signaling functions of these adhesion proteins have been found to be important in embryonic development and formation of the nervous system and other tissues, in holding tissues together in adults, in inflammation and wound healing, and in the metastasis of tumors. Many CAMs pass through the cell membrane and are anchored to the cytoskeleton inside the cell. Some bind to like molecules on other cells (homophilic binding), whereas others bind to nonself molecules (heterophilic binding). Many bind to laminins, a family of large cross-shaped molecules with multiple receptor domains in the extracellular matrix.
CLINICAL BOX 2–3 Ciliary Diseases
Primary ciliary dyskinesia refers to a set of inherited disorders that limit ciliary structure and/or function. Disorders associated with ciliary dysfunction have long been recognized in the conducting airway. Altered ciliary function in the conducting airway can slow the mucociliary escalator and result in airway obstruction and increased infection. Dysregulation of ciliary function in sperm cells has also been well characterized to result in loss of motility and infertility. Ciliary defects in the function or structure of primary cilia have been shown to have effects on a variety of tissues/organs. As would be expected, such diseases are quite varied in their presentation, largely due to the affected tissue, and include mental retardation, retinal blindness, obesity, polycystic kidney disease, liver fibrosis, ataxia, and some forms of cancer.
THERAPEUTIC HIGHLIGHTSThe severity in ciliary disorders can vary widely, and treatments targeted to individual organs also vary. Treatment of ciliary dyskinesia in the conducting airway is focused on keeping the airways clear and free of infection. Strategies include routine washing and suctioning of the sinus cavities and ear canals and liberal use of antibiotics. Other treatments that keep the airway from being obstructed (eg, bronchodilators, mucolytics, and corticosteroids) are also commonly used.
Nomenclature in the CAM field is somewhat chaotic, partly because the field is growing so rapidly and partly because of the extensive use of acronyms, as in other areas of modern biology. However, the CAMs can be divided into four broad families: (1) integrins, heterodimers that bind to various receptors; (2) adhesion molecules of the IgG superfamily of immunoglobulins; (3) cadherins, Ca2+-dependent molecules that mediate cell-to-cell adhesion by homophilic reactions; and (4) selectins, which have lectin-like domains that bind carbohydrates. Specific functions of some of these molecules are addressed in later chapters.
The CAMs not only fasten cells to their neighbors, but they also transmit signals into and out of the cell. For example, cells that lose their contact with the extracellular matrix via integrins have a higher rate of apoptosis than anchored cells, and interactions between integrins and the cytoskeleton are involved in cell movement.
Intercellular junctions that form between the cells in tissues can be broadly split into two groups: junctions that fasten the cells to one another and to surrounding tissues, and junctions that permit transfer of ions and other molecules from one cell to another. The types of junctions that tie cells together and endow tissues with strength and stability include tight junctions, which are also known as the zonula occludens (Figure 2–8). The desmosome and zonula adherens also help hold cells together, and the hemidesmosome and focal adhesions attach cells to their basal laminas. The gap junction forms a cytoplasmic “tunnel” for diffusion of small molecules (< 1000 Da) between two neighboring cells.
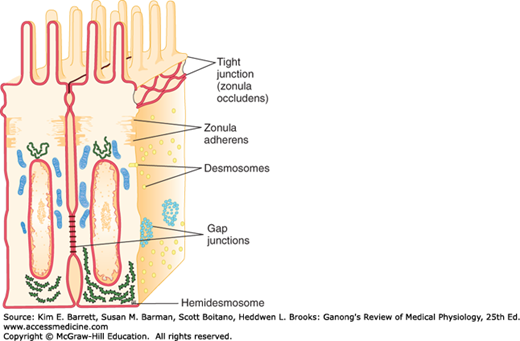
Tight junctions characteristically surround the apical margins of the cells in epithelia such as the intestinal mucosa, the walls of the renal tubules, and the choroid plexus. They are also important to endothelial barrier function. They are made up of ridges—half from one cell and half from the other—which adhere so strongly at cell junctions that they almost obliterate the space between the cells. There are three main families of transmembrane proteins that contribute to tight junctions: occludin, junctional adhesion molecules, and claudins; there are several more proteins that interact from the cytosolic side. Tight junctions permit the passage of some ions and solute in between adjacent cells (paracellular pathway) and the degree of this “leakiness” varies, depending in part on the protein makeup of the tight junction. Extracellular fluxes of ions and solute across epithelia at these junctions are a significant part of overall ion and solute flux. In addition, tight junctions prevent the movement of proteins in the plane of the membrane, helping maintain the different distribution of transporters and channels in the apical and basolateral cell membranes that make transport across epithelia possible.
In epithelial cells, each zonula adherens is usually a continuous structure on the basal side of the zonula occludens, and it is a major site of attachment for intracellular microfilaments. It contains cadherins.
Desmosomes are patches characterized by apposed thickenings of the membranes of two adjacent cells. Attached to the thickened area in each cell are intermediate filaments, some running parallel to the membrane and others radiating away from it. Between the two membrane thickenings, the intercellular space contains filamentous material that includes cadherins and the extracellular portions of several other transmembrane proteins.
Hemidesmosomes look like half-desmosomes that attach cells to the underlying basal lamina and are connected intracellularly to intermediate filaments. However, they contain integrins rather than cadherins. Focal adhesions also attach cells to their basal laminas. As noted previously, they are labile structures associated with actin filaments inside the cell, and they play an important role in cell movement.
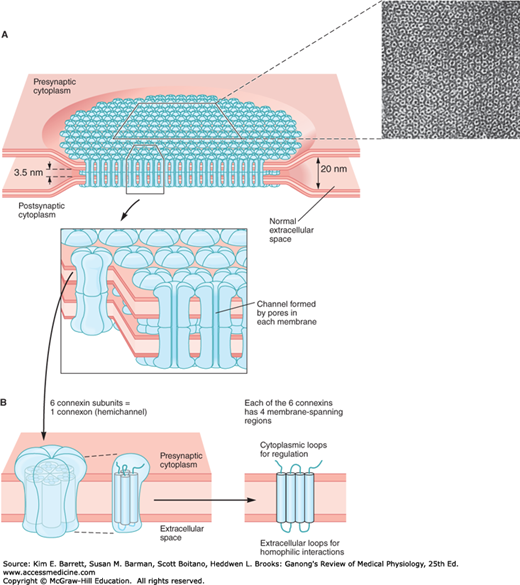
The intercellular space is up to 4 nm at gap junctions. Here, units called connexons in the membrane of each cell are lined up with one another to form the dodecameric gap junction (Figure 2–9). Each connexon is made up of six protein subunits called connexins. They surround a channel that, when lined up with the channel in the corresponding connexon in the adjacent cell, permits substances to pass between the cells without entering the extracellular fluid (ECF). The pore diameter in the channel is estimated between 0.8 and 1.4 nm, which permits the passage of ions, sugars, amino acids, and other solutes with molecular weights up to about 1000 Da. Gap junctions thus permit the rapid propagation of electrical activity from cell to cell, as well as the exchange of various chemical messengers. However, the gap junction channels are not simply passive, nonspecific conduits. At least 20 different genes code for connexins in humans, and mutations in these genes can lead to diseases that are highly selective in terms of the tissues involved and the type of communication between cells produced (Clinical Box 2–4). Experiments in which particular connexins are deleted by gene manipulation or replaced with different connexins confirm that the particular connexin subunits that make up connexons determine their permeability and selectivity. It should be noted that connexons can also provide a conduit for regulated passage of small molecules between the cytoplasm and the ECF. Such movement can allow additional signaling pathways between and among cells in a tissue.
FIGURE 2–9
Gap junction connecting the cytoplasm of two cells. A) A gap junction plaque, or collection of individual gap junctions, isshown to form multiple pores between cells that allow for the transfer of small molecules. Inset is an electron micrograph from rat liver. B) Topographic depiction of individual connexon and corresponding six connexin proteins that traverse the membrane. Note that each connexin traverses the membrane four times. (Reproduced with permission from Kandel ER, Schwartz JH, Jessell TM, Siegelbaum SA, Hudspeth AJ (editors): Principles of Neural Science, 5th ed. New York, NY: McGraw-Hill; 2013.)
A nucleus is present in all eukaryotic cells that divide. The nucleus is made up in large part of the chromosomes, the structures in the nucleus that carry a complete blueprint for all the heritable species and individual characteristics of the animal. Except in germ cells, the chromosomes occur in pairs, one originally from each parent. Each chromosome is made up of a giant molecule of DNA. The DNA strand is about 2 m long, but it can fit in the nucleus because at intervals it is wrapped around a core of histone proteins to form a nucleosome. There are about 25 million nucleosomes in each nucleus. Thus, the structure of the chromosomes has been likened to a string of beads. The beads are the nucleosomes, and the linker DNA between them is the string. The whole complex of DNA and proteins is called chromatin. During cell division, the coiling around histones is loosened, probably by acetylation of the histones, and pairs of chromosomes become visible, but between cell divisions only clumps of chromatin can be discerned in the nucleus. The ultimate units of heredity are the genes on the chromosomes. As discussed in Chapter 1, each gene is a portion of the DNA molecule.
CLINICAL BOX 2–4 Connexins in Disease
There is extensive information related to the in vivo functions of connexins, growing out of work on connexin knockouts in mice and the analysis of mutations in human connexins. The mouse knockouts demonstrated that connexin deletions lead to electrophysiologic defects in the heart and predisposition to sudden cardiac death, female sterility, abnormal bone development, abnormal growth in the liver, cataracts, hearing loss, and a host of other abnormalities. Information from these and other studies has allowed for the identification of several connexin mutations known to be responsible for almost 20 different human diseases. These diseases include several skin disorders such as Clouston syndrome (a connexin 30 (Cx30) defect) and erythrokeratoderma variabilis (Cx30.3 and Cx31); inherited deafness (Cx26, Cx30, and Cx31); predisposition to myoclonic epilepsy (Cx36); predisposition to arteriosclerosis (Cx37); cataract (Cx46 and Cx50); idiopathic atrial fibrillation (Cx40); and X-linked Charcot-Marie-Tooth disease (Cx32). It is interesting to note that each of these target tissues for disease contain other connexins that do not fully compensate for loss of the crucial connexins in disease development. Understanding how loss of individual connexins alters cell physiology to contribute to these and other human diseases is an area of intense research.
The nucleus of most cells contains a nucleolus (Figure 2–1), a patchwork of granules rich in RNA. In some cells, the nucleus contains several of these structures. Nucleoli are most prominent and numerous in growing cells. They are the site of synthesis of ribosomes, the structures in the cytoplasm in which proteins are synthesized.
The interior of the nucleus has a skeleton of fine filaments that are attached to the nuclear membrane, or envelope (Figure 2–1), which surrounds the nucleus. This membrane is a double membrane, and spaces between the twofolds are called perinuclear cisterns. The membrane is permeable only to small molecules. However, it contains nuclear pore complexes. Each complex has eightfold symmetry and is made up of about 100 proteins organized to form a tunnel through which transport of proteins and mRNA occurs. There are many transport pathways; many proteins that participate in these pathways, including importins and exportins, have been isolated and characterized. Much current research is focused on transport into and out of the nucleus, and a more detailed understanding of these processes should emerge in the near future.
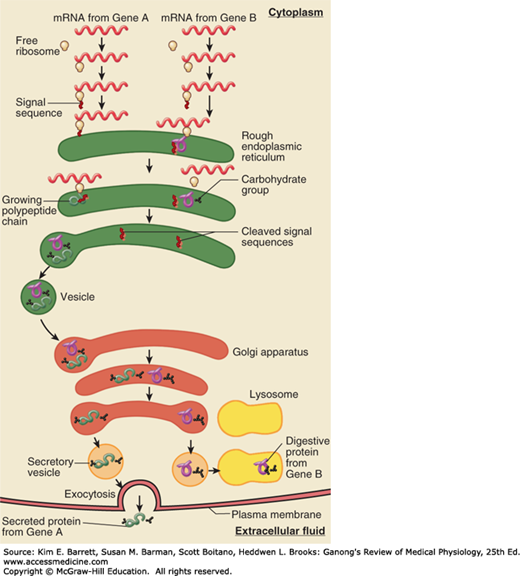
The endoplasmic reticulum is a complex series of tubules in the cytoplasm of the cell (Figure 2–1; Figure 2–10; and Figure 2–11). The inner limb of its membrane is continuous with a segment of the nuclear membrane, so in effect this part of the nuclear membrane is a cistern of the endoplasmic reticulum. The tubule walls are made up of membrane. In rough (granular) endoplasmic reticulum, ribosomes are attached to the cytoplasmic side of the membrane, whereas in smooth (agranular) endoplasmic reticulum, ribosomes are absent. Free ribosomes are also found in the cytoplasm. The granular endoplasmic reticulum is concerned with protein synthesis and the initial folding of polypeptide chains with the formation of disulfide bonds. The agranular endoplasmic reticulum is the site of steroid synthesis in steroid-secreting cells and the site of detoxification processes in other cells. A modified endoplasmic reticulum, the sarcoplasmic reticulum, plays an important role in skeletal and cardiac muscle. In particular, the endoplasmic or sarcoplasmic reticulum can sequester Ca2+ ions and allow for their release as signaling molecules in the cytosol.
FIGURE 2–10
Rough endoplasmic reticulum and protein translation. Messenger RNA and ribosomes meet up in the cytosol for translation. Proteins that have appropriate signal peptides begin translation, and then associate with the endoplasmic reticulum (ER) to complete translation. The association of ribosomes is what gives the ER its “rough” appearance. (Reproduced with permission from Widmaier EP, Raff H, Strang KT: Vander’s Human Physiology: The Mechanisms of Body Function, 11th ed. New York, NY: McGraw-Hill; 2008.)