Fig. 4.1
The effect of a hydrophilic diluent on the dissolution rate of a sparingly soluble, hydrophobic active substance in a gelatine capsule (from McConell and Basit [7], with permission).
a–c: Gelatine capsule with only hydrophobic active substance particles.
d–g: Gelatine capsules with hydrophobic active substance particles and a hydrophilic, in water-soluble diluent
Problems may arise in the presence of hydrophobic active substances or excipients that are not wetted easily. In these cases it may be necessary to add a disintegrating agent to the formulation.
The choice of a diluent may influence the absorption of an active substance, which was seen in the 1960s in Australia. The diluent of phenytoin sodium capsules was changed from calcium sulfate dihydrate to lactose, which strongly enhanced the bioavailability of phenytoin sodium. Plasma levels of phenytoin increased up to fourfold, which led to an increased reporting of adverse events [8, 9]. This case drew worldwide attention and resulted in an increased awareness of the importance of pharmaceutical availability and bioavailability of active substances in the development of solid oral dosage forms.
Capsules release their contents when at least a part of the capsule shell is dissolved. The Ph. Eur. requires that capsules disintegrate within 30 min [6]. However, the shell of gelatine capsules usually dissolves within 3–15 min in the aqueous, acidic gastric lumen. The powder in capsules prepared in the pharmacy has not been subjected to a compression stage, as is the case of most industrially manufactured capsules. Therefore, the content of pharmacy prepared capsules is usually released more quickly than industrial prepared capsules.
Oral powders (single-dose or multidose) and the contents of opened capsules do not require the release of the active substance from the dosage form. Therefore, only the dissolution rate of the active substance itself is important, provided no agglomeration is observed and the crystalline structure of the active substance has not changed during manipulation and exposure to air. Consequently, pharmaceutical availability and absorption rate of sparingly soluble or slowly dissolving powders are comparable to those of oral suspensions. Effervescent powders and powders that dissolve well in water (preferably dissolved prior to ingestion) have a bioavailability which is (almost) equal to oral solutions.
Tablets are compressed preparations, and therefore, require a disintegrating agent to promote their disintegration by swelling, dissolving or becoming effervescent in contact with water. Furthermore, the hardness of a tablet is important. The Ph. Eur. requires that non-coated tablets disintegrate within 15 min in water. Currently available disintegrating agents allow the preparation of tablets which disintegrate within a few minutes.
In addition to the disintegration of a capsule or tablet, absorption rate is determined by the dissolution rate of the active substance. The dissolution rate of the active substance depends on various factors, for example the solubility, the particle size and shape, the crystal morphology, and wetting ability. Section 18.1 discusses the effects of these factors on the absorption of the active substance.
Ingestion of a tablet should not always lead to a rapid release. In a number of cases it may be preferred that the active substance is not released directly, for example [10]:
The active substance degrades in gastric juice, or irritates the stomach wall (e.g. valproic acid).
The active substance should exert its effect in a specific part of the intestine or should reach undamaged a specific part of the intestine for absorption (e.g. mesalazine).
Absorption of the active substance should be spread out evenly over a period of time to reach an appropriate plasma concentration (e.g. morphine, theophylline).
The therapeutic benefits from a specific release pattern over time (e.g. methylphenidate, for which it may be therapeutically relevant to have an immediate release of a small fraction of the dose whereas the largest fraction is controlled released).
The patient benefits from a less frequent dosing regimen, i.e. the release occurs over an extended period of time (e.g. clomipramine). Modified-release tablets or enteric-coating tablets do not release the active substance directly, but do so in a specific part of the gastro-intestinal tract, either delayed or with a specific release pattern. The release of these tablets is adjusted to therapeutic needs of the patient.
Pentasa® microgranules are an example of a dosage form designed to release the active substance (mesalazine) in a specific part of the intestines. Pentasa® tablets disintegrate into microgranules following oral administration, whilst Pentasa® sachets contain the microgranules as such. The release rate of mesalazine from the microcapsules is pH dependent (faster release at higher pH), which results in a continuous release of mesalazine in the small and large intestines at all enteral pH conditions. After one 1,500 mg dose, approximately 60 % is released in the small intestine, and 40 % is released in the large intestine. Mesalazine is partly metabolised by the intestinal mucosa to acetylmesalazine. About 30 % of the ingested dose is absorbed in the small intestine, and 25 % in the colon (predominantly as acetylmesalazine) [11].
4.4 Product Formulation
The design of the formulation of capsules and oral powders and that of tablets are similar in some respects, but there are also some important differences. In this section, the general aspects of formulation design are discussed.
4.4.1 The Need for Excipients
It is usually not possible to prepare a capsule, oral powder or tablet from an active substance without the addition of any excipients. Firstly, the volume of the active substance is often very small; a diluent is necessary in order to handle the powder mixture. Secondly, the active substance may not have good flow properties; these can be improved by addition of a glidant. Another reason to use excipients is that a preparation, consisting only of an active substance, may not disintegrate well in the gastro-intestinal tract; a disintegrating agent can improve this. Many excipients combine a number of such functions so the number of different excipients can be limited and the potential interactions between materials can be minimised [12]. The next sections discuss these functions in relation to the required properties of solid oral dosage forms.
The intrinsic properties of the active substance are difficult to change, but the pharmacist can choose the right excipients and preparation techniques to overcome or decrease the impact of limitations. Although excipients should be pharmacologically inactive, they may cause adverse effects. The European Paediatric Formulation Initiative (EuPFI) project is considering the suitability of excipients for paediatric formulations. The results have been published in the STEP database [13]. For example many colouring agents have been associated with hypersensitivity and other adverse reactions.
4.4.2 Active Substance
Solid oral dosage forms are preferably prepared with the active substance as such. The particle size of active substances in fast-release preparations should preferably be not larger than 180 μm to reach a compromise between dissolution rate and flowability. If the raw material consists of particles that are too large, the particles should be reduced in size (see Sect. 29.2).
When the active substance is not available as raw material, tablets containing the active substance may be used, providing that both the tablet and the active substance are suitable for processing into a capsule. Sometimes the active substance is extracted by dissolution into a liquid but unfortunately these solutions (especially aqueous liquids) cannot be further used in the preparation of capsules because they affect the gelatine shell. However, there are exceptions such as macrogols of small chain lengths [14], which do not affect the gelatine.
Any processing of the active substances (e.g. milling, hydration), which may occur during preparation, can modify their physical properties. This is probably not being noticed by the pharmacist as he will not have methods at his disposal to confirm changes of the active substances [15]. For this reason, preparation of dosage forms should preferably be carried out starting from the raw material of which the quality meets the requirements. Thus, the availability of pure qualified active substances is advantageous for the preparation of adapted doses and reduces the risk of manipulations of licensed products.
Highly soluble salts (for example sodium fluoride, potassium chloride, potassium citrate) are preferably not prepared in a capsule at all, since rapid dissolution can result in a high local concentration that may be harmful to the mucosa of the gastro-intestinal tract. An enteric coating on capsules and tablets can protect the gastric mucosa from irritating active substances. But the preparation of an oral solution of the active substance may be a better alternative.
4.4.3 Dilution and Flowability of the Powder Mixture
Solid oral divided dosage forms are prepared by dividing a mixture of the active substance and excipients evenly over a dosing mould, so every unit corresponds to one dose. In the case of capsules or oral powders, the powder is spread over the capsule shells or powder papers, respectively. Moulds should be filled evenly. Therefore, good flowability is required.
The flowability of a powder (mixture) can be tested in several ways but a relevant impression for small-scale preparation is obtained from observation during mixing [16]. When the mixture is dusty, sticky, or when segregation of components occurs, other excipients should be used or a glidant has to be added. Even if the mixture looks all right, powder flow may not be good enough. Insufficient flow will lead to uneven filling of the separate capsules and tablet moulds, and ultimately result in too large a weight variation. By preparing trial batches with various filling agents and glidants, and comparing the weight distributions, the powder formulation with the best flow properties can be selected. Flowability of powder can be measured directly (flow through an orifice) or indirectly (angle of repose or tapped and bulk densities) [17].
Powder flowability is influenced by [18]:
The particle size: powders consisting of many small particles tend to show a poor flow.
The particle shape: a more regular shape promotes good flow properties, particularly the spherical shape.
The surface of the particle: a smoother surface results in better flow properties. Furthermore, the surface of particles can be modified to improve the flow properties.
The moisture content of the powder (this can vary under ambient conditions): the powder flows better when the moisture content is low, but too low will generate static electricity.
Electrostatic charge: removing the charge improves the flow properties.
4.4.3.1 Diluents
Addition of excipients such as a diluent with good flow properties may improve the flow properties of a powder. Diluents are added to powder mixtures also to increase the mass and volume of the active substance. Very small amounts of active substances often require a carrier to ensure their uniform distribution in the dispensed product, and to guarantee an accurate dose [19].
The most often used diluent for capsules and powders is microcrystalline cellulose (Avicel PH 102 or Pharmacel 102, see also Sect. 23.4.1). Microcrystalline cellulose has both proper flow and disintegrating properties. However, microcrystalline cellulose has some drawbacks. For instance it is insoluble in water forming a suspension. Secondly, the active substance may adsorb onto cellulose particles, which may reduce both solubility and dissolution rates of sparingly water soluble active substances, and thereby the active substance’s relative pharmaceutical bioavailability. Microcrystalline cellulose causes no systemic adverse effects, because humans do not absorb it [12, 20].
Lactose (alfa-lactose monohydrate) (see also Sect. 23.4.4) has somewhat less favourable flow properties than microcrystalline cellulose PH102. A disadvantage of lactose is its incompatibility with primary amines. An advantage compared to microcrystalline cellulose is that it is water soluble, which makes lactose suitable for capsules where the contents have to be dissolved. Capsules containing lactose disintegrate as a result of the dissolution of lactose (Fig. 4.1). Its use might be limited in patients with lactose intolerance [12].
Dried (corn, rice or potato) starch (see also Sect. 23.4.1) has good flow and disintegrating properties. It is used occasionally as a diluent in capsules for the processing of hygroscopic substances. Starch is extracted from plant material and subsequently dried. The water content should be below 5 %. During a few hours of exposure to air with a relative humidity of about 60 %, dried starch will take up 5 % of water.
4.4.3.2 Glidants
If not enough diluent is present, or if the powder does not flow sufficiently despite the presence of a relatively large quantity of diluent, the addition of a glidant can be considered. When a glidant is required for the preparation of capsules, preferably colloidal anhydrous silica (Aerosil 200 V) is used in a fraction of 0.5 %. However, it has a tendency to adsorb onto active substance particles, so the application should be investigated beforehand. Magnesium stearate can be used as an alternative, but it is not preferred because its hydrophobic nature may negatively influence the wetting and dissolution rate of the active substance.
A glidant does not always improve the powder flowability, for example when the active substance is micronised. The cohesion forces between the small particles may be too large to be overcome by a glidant. Moreover, when the poor flow properties of the powder are due to irregular particle shapes, a glidant will not have much effect either.
Addition of a glidant could be counterproductive because of [21]:
Segregation: glidants may displace the active substance particles that are bound to a carrier by ordered mixing (see Sect. 4.5.1). The small particles that are displaced may decrease the flowability and the powder mixture may segregate.
Segregation and loss: a glidant may also increase the flowability too much. Small particles may move too easily between the large particles, which may lead to segregation of the powder with the large particles on top and the small particles at the bottom. Moreover, particles may fall between the capsule shell and the capsule filling apparatus, resulting in loss of active substance content.
Incompatibilities: glidants may cause degradation of other substances. For example, magnesium stearate may react with acids.
Reduced dissolution rate: magnesium stearate is hydrophobic and forms a hydrophobic layer on the surface of the powder particles. Therefore, the dissolution rate of the active substance may be reduced.
Reduced pharmaceutical availability: colloidal silicon dioxide has a large surface area, which may facilitate adsorption to the active substance. This may reduce the pharmaceutical availability of the active substance.
4.4.3.3 Binding Agents
Binding agents combine the diluent function – and thus improve flowability – and the binding function mainly used in direct compression of tablets. These excipients increase the mass and promote the bonds between particles of other materials in the formulation, so in fact they lead to, desired, agglomeration. In direct compression the powder mixture is not granulated before compression. Therefore, binding agents should improve flowability without segregation of the mixture.
In direct compression tablets, microcrystalline cellulose of various grades is used as a binding agent. Generally the PH101 grade with a mean particle size of 50 μm and the PH102 grade with a mean particle size of 90 μm are used. The PH101 grade flows poorly, not only because of the small particle size, but also because of the needle like particle shape. The PH102 quality flows better because half of the particles are granulated.
Calcium monohydrogen phosphate dihydrate is used in granulated grade as a binding agent in tablets prepared by direct compression. Since the binding properties are quite poor, it is usually combined with another binding agent. In capsules calcium monohydrogen phosphate dihydrate is used when none of the common diluents are suitable, for example for the processing of corticosteroids (Table 4.1). In spite of its hydrophilic nature calcium monohydrogen phosphate dihydrate has neither disintegrating properties, nor it is water-soluble, therefore, addition of a disintegrating agent is required. Primojel Capsule diluent FNA is a diluent for capsules that contains, besides calcium monohydrogen phosphate dihydrate, the disintegrating agent sodium starch glycolate A (Primojel®) and the glidant silica colloidal anhydrous (Table 4.2).
Calcium hydrogen phosphate dihydrate, heavya | 94 g |
Silica, colloidal anhydrous compressed | 1 g |
Sodium starch glycolate (type A)b | 5 g |
Total | 100 g |
During an investigation into the optimal formulation of a well flowing powder for the preparation of Prednisolone capsules, microcrystalline cellulose with anhydrous colloidal silica failed to give a mean content meeting the requirements [22]. Apparently the electric charge was not neutralised and prednisolone was lost through flying up and through the exhaust. Calcium monohydrogen phosphate dihydrate in combination with colloidal silicon dioxide gave the best results. However, due to the lack of disintegrating properties of calcium monohydrogen phosphate dihydrate, a solid mass remained after dissolving of the capsule shell. Only capsules with prednisolone, calcium monohydrogen phosphate dihydrate, silicon dioxide and the disintegrating agent sodium starch glycolate disintegrated giving a desired dissolution rate.
4.4.4 Disintegration
Capsules disintegrate when the capsule shell dissolves and the powder mixture is wetted. Hydrophilic excipients promote the wetting of the powder bed (Fig. 4.1). Due to the low compaction of the encapsulated powder, and the easy dissolution of most diluents for capsules, the addition of a disintegrating agent is often not needed for pharmacy preparations. However, when excipients compact easily (e.g. calcium monohydrogen phosphate dihydrate) a disintegrant is recommended.
Disintegrating agents act through swelling or by promoting water penetration through capillary action or even by the production of a gas (e.g. effervescence) (see Fig. 4.1). Highly compressed powders or granulates in tablets are more difficult to disintegrate, thus, the addition of a disintegrating agent is often required for immediate release of the active substance.
The diluents microcrystalline cellulose and lactose have some disintegrating properties. When these diluents are used in capsules, the addition of a separate disintegrating agent may not be necessary, or it is used only in smaller fraction than in tablets. However, when a diluent without disintegrating properties is used, such as calcium monohydrogen phosphate dihydrate, a disintegrating agent has to be added, particularly 5 % of sodium starch glycolate [23]. For instance sodium starch glycolate exerts its disintegrating effect by strongly swelling in the presence of water, which leads to the breaking of bonds in the powder bed or tablet. Lactose disintegrates powder beds by dissolution in water. However, tablets for immediate release of active substances always contain a disintegrating agent.
4.4.5 Incompatibilities
The most important incompatibility in capsules is the adsorption of active substances to excipients and vice versa. Sparingly water-soluble active substances may adsorb to non-water soluble excipients such as microcrystalline cellulose (diluent). On the other hand, the very fine glidant, colloidal anhydrous silica, can adsorb onto active substance particles. Especially for low dosed active substances, relatively large fractions may adsorb or be adsorbed. Such adsorption may delay the dissolution of the active substance, resulting in a delayed or incomplete release of the substance. This may lead to a reduced pharmaceutical availability and ultimately a lower therapeutic activity. Substances known to adsorb to microcrystalline cellulose are ethinylestradiol and dexamethasone [24].
Due to the absence of water, which catalyses many chemical reactions, chemical incompatibilities rarely occur in dry dosage forms. One exception is the incompatibility of the excipient lactose with primary amines, such as amphetamine and lisinopril. The rate of the reaction (Maillard reaction) is slow in absence of water, but may lead to yellow discolouration during storage [25].
Thus the pharmacist must be careful when choosing the excipients. Pre-formulation studies to identify incompatibilities can be time-consuming but are required to prove that no instability of the active substance will occur during preparation and storage. In the case of the use of licensed products to prepare capsules or powders, it might be difficult to obtain information from the manufacturer. Thus, it is usually safe to dilute crushed tablet with the excipient that is used in the tablet formulation, based on the market authorisation holder who has ensured their compatibility.
4.4.6 Colouring and Flavouring
A capsule or tablet that is swallowed as such is almost tasteless, because only a small part of the active substance comes into contact with the palate. Therefore taste masking is generally not necessary. If taste masking is required for tablets, a coating can be applied. Patients who cannot swallow capsules receive either a powder or the contents of a capsule. The direct contact between powder and palate results in a distinct taste sensation. Many active substances have an unpleasant taste and thus flavouring, sweetening and even colouring of the powder are vital to patient compliance. Taking the powder with food – e.g. yoghurt or custard – can also be an adequate way to mask the taste, particularly for children, which are very sensitive to organoleptic properties. One of the scopes of a large European research project on children’s medicines is to discover more about masking taste and smell in oral dosage forms [26].
One cannot simply add a flavour to a dosage form containing a unpleasent tasting active substance and expect it to taste good because the strength of tastes are different or different receptors are sensitised. Flavours are complex mixtures that are made up of many chemicals. Flavouring agents can be natural (essential oil, derivatives from fruit vegetable juice) or artificial (see Sect. 5.4.10). In particularly, natural flavours may have dozens of different types of molecules, which may interfere with the active substance.
In some cases it may be desirable to colour the tablet or capsule, for example to prevent a mix-up of medicines or to prepare placebos with an appearance identical to the original tablets for use in clinical trials. In case of capsules, it is possible to use coloured capsule shells, or to prepare a coloured powder mixture in transparent capsules. Colouring agents should be used with caution, because they can cause allergic reactions. The use of coloured capsule shells is preferred, but if it is necessary to colour the powder mixture, a colouring agent can be added. Section 23.11 lists colouring agents for powder mixtures. Tablets can be coloured by using soluble (for wet granulation) or insoluble (for direct compression) colouring agents. Moreover, tablets are often coloured by processing of a colouring agent in the coating.
Patients with Addison’s disease or Cushing’s syndrome take steroids two or three times a day in various doses, depending on the time of the day and the situation. Commercially available tablets may not always contain the dose they need. Moreover, tablets with different doses are not always easy to distinguish. For these patients, capsules with a dose-related colour can be a solution [27].
4.5 Method of Preparation
The preparation of solid oral dosage forms consists of two steps. The first step is the preparation of a homogeneous powder mixture, and the second step is the even distribution of the powder mixture over the dose units. Mixing of solids to obtain a homogeneous mixture is in principle the same process whether capsules, powders or tablets are prepared. However, the requirements regarding the filling of the dose units are different for the three types of preparation.
Mass for oral powders is easy to prepare but time-consuming to divide. The solids are mixed together and subsequently the powder mixture is divided evenly over the powder papers. The same applies to cachets. The preparation of capsules is quick but somewhat more complex, because the powder mixture should have a fixed volume, which is determined beforehand. Next, the powder mixture has to be divided evenly over the capsule shells. The preparation of tablets is in this regard more complex. Tablets are made with a tableting machine (see Sect. 28.7.3 for some brands), which imposes extra requirements to the flowability of the powder mixture. To minimise flow and segregation problems, powder mixtures are often granulated before compression.
4.5.1 Homogeneous Powder Mixtures
4.5.1.1 Particle Size
Particle size of the constituents for powders range commonly from 10 μm up to 180 μm. For the preparation of a homogeneous mixture, the solids preferably have comparable particle sizes, densities, shapes and equal mixing ratio (see Sect. 4.4.2). Particles with unequal sizes mix poorly and may segregate easily. Segregation, for example during the filling of the capsules, may lead to a large weight variation and a bad content uniformity. In practice, the maximum particle size for the active substance is 180 μm, unless a delayed release effect is envisaged. Larger particles have a relatively small surface area, which may result in a too low dissolution rate (see Sect. 18.1). When the particles of the starting material are larger than 180 μm, the material should be ground and, if necessary, sieved. However, grinding by hand (triturating) of materials that are already fine enough should be avoided, because it may introduce agglomerates.
4.5.1.2 Starting from Tablets or Capsules
Real challenges for the preparation of the powder mixture arise from the situation in which tablets or capsules are needed to get access to the active substance. In the first place the pharmacist has to consider the suitability for grinding or crushing of the tablets or capsules and then he has to develop a reliable method to produce the required dilution of the ground mass; this dilution sometimes having to be quite considerable, even a 100-fold.
Coated tablets, such as enteric-coated tablets, and modified-release tablets are better not split or pulverised, because their specific features may be lost. If it is absolutely necessary to break them then the pharmacist must know beforehand the implications on the stability of the medicine and on its therapeutic effect (see Sect. 4.9). A controlled-release tablet that has been split may overdose. Splitting may also expose the taste of the medicine, which had originally been masked in the coated tablet. Only standard, non-coated tablets shall readily be processed into a powder mixture.
To obtain the required amount of active substance the equivalent amount of whole tablets are counted. It is preferable to use several tablets to level out content differences between tablets. In principle there are two ways: take an exact, counted number of tablets to pulverise, or to use an excess of tablets in pulverising and then weighing the required quantity. If an exact number of tablets is used, the resulting mean content of active substance in the final product has to be validated.
A strict method of grinding is needed to avoid the loss of active substance for example due to static charges or to sticking of tablet components to utensils such as mortar, pestle and tablet crusher device. This loss of active substance can be compensated beforehand by taking an excess of tablets. Note, however, the risk of calculation errors in that case. There is some loss of active substance also during administration, i.e. taking the powder from folded paper or emptying the capsule, and the loss seems to be higher in low-weight single-dose powders (Fig. 4.2).
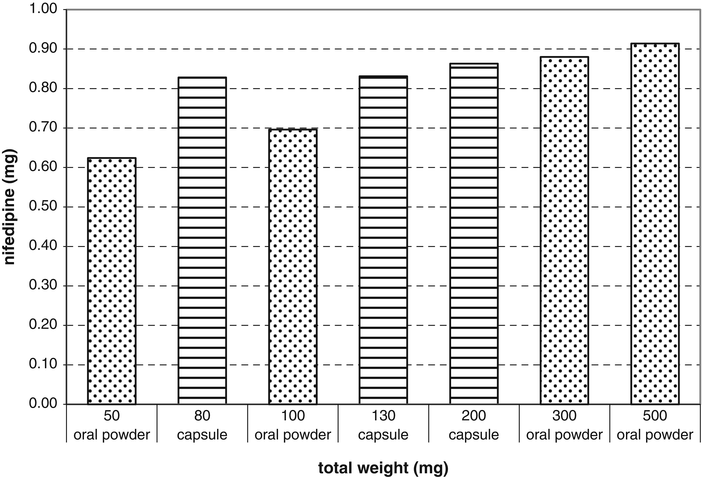
Fig. 4.2
Nifedipine contents in single-dose powders or capsules that were meant to contain nifedipine 1 mg as active substance (from crushed tablets Adalat® 10 mg retard) and microcrystalline cellulose (MCC) as a diluent. The batch size was 50 single-dose powder or capsules and no excess tablets were used. Capsules number 4 (80 mg MCC), 3 (130 mg MCC) and 1 (200 mg MCC) are compared to oral powders weighing 50 mg, 100 mg, 300 mg and 500 mg. Mean values are shown (n = 10). The higher content of nifedipine can be observed in all sizes of capsules compared to oral powders of 50 mg or 100 mg. According to this study the use of different sizes of capsules would lead to an acceptable content while oral powders should weigh at least 300 mg [28]
Tablets are crushed manually in a non-porous mortar with a pestle. After careful grinding the resulting powder may be more or less homogeneous (Fig. 4.3).

Fig. 4.3
Particle size variation in the manually (in a mortar with a pestle) crushed Nifedipine tablet (Adalat® 10 mg retard). Scale bar in SEM is 100 μm [29]
The properties of all excipients present in the tablet must be considered for their possible effects on the final preparation, such as weight variation, disintegration time, dissolution characteristics and in vivo performance. The lower the proportion of the active substance present in the mixture, the more difficult it is to achieve a sufficient homogeneity. In Sects. 4.9 and 4.10, the formulation of tablets and the possibilities to process coated or modified-release tablets in capsules are discussed.
4.5.1.3 The Mixing Process
Geometric dilution is used to ensure that small quantities of ingredients are uniformly distributed in the powder mixture, starting with the ingredient in the smallest quantity. Then a volume of powder equal to the volume of the powder mixture in the mortar is added and triturated with a pestle to a uniform mixture. Then the mixture of the two components is mixed again with an equal quantity of diluent. This process is repeated until all solids are mixed. Triturating and small-scale mixing is performed in a mortar with pestle (Sect. 28.6.4). For larger quantities a mixing apparatus needs to be used.
Mixing should be performed in such a way that the following problems are avoided:
1.
Adsorption of the active substance to the pestle, mortar, measuring cylinder or the mixer. Adsorption of active substances may be reduced by minimising direct contact with utensils. Therefore, the active substance is best put in between the utensils and the excipient (‘wrapping method’, see Sect. 4.6.2). Mortars need to be non-porous so that no active substance remains in the pores to decrease the dose or to contaminate the next product to be prepared.
2.
Suctioning of the statically charged active substance with the airflow, since mixing of powders is generally performed in the presence of dust extraction. If it is assumed that substances are selectively suctioned when they become statically charged, this can be avoided by selecting excipients (e.g. colloidal anhydrous silica) that can neutralise the static charge, by reducing the mixing time to the minimum, and by avoiding whipping too much air into the mixture. A powder that is too fluffy can be compacted slightly to change the surface properties by the addition of a few drops of alcohol, water or liquid paraffin.
In the past, homogeneity of powder mixtures was assessed by processing a colouring agent in the powder mixture. It was assumed that when the colour was spread homogeneously over the mixture, the entire mixture was homogeneous. However, this method may not be suitable for two reasons. First, a homogeneous distribution of a colouring agent is not a guarantee that the active substance is distributed evenly over the mixture as well. The physical properties of solids, such as particle size and adsorption onto other substances, determine the quality of the mixing. Differences in properties of colouring agent and active substance may result in a different distribution for both substances. A second reason is that colouring agents may cause hypersensitivity.
4.5.1.4 Solvent Method
Mixing of powders with unequal volumes often results in non-homogeneous mixtures, because it takes more patience to obtain a homogeneous mixture than when two equal parts are mixed. However, small amounts of active substances simply need to be mixed with a large quantity of diluent to obtain a processable powder mixture. In case of an inconvenient mixing ratio, the solvent method might be applicable, if validated carefully. The method has to be investigated for a specific active substance and standardised formulation. Unfortunately the solvent method often appears not to be appropriate because there is no suitable solvent, or because the active substance is not stable in solution.
The solvent method basically distributes a small quantity of active substance (5 mg or less per dose unit) over a diluent. The active substance is dissolved in a suitable, volatile solvent, usually in a stainless steel mortar to be able to check if dissolution is completed. -Subsequently, the solution is mixed with a carrier that does not dissolve in the solvent. The moistened powder is triturated –until the solvent has completely evaporated. The powder mixture now consists of carrier particles with a coating of the active substance. This method is in fact a combination of simultaneous particle size reduction and mixing. Another advantage of this method is the reduced loss of the powder mixture during mixing.
The solvent should comply with a number of requirements:
The active substance has to dissolve well in the solvent, but not the carrier.
The solvent has to be volatile enough in order that the powder will dry within a limited period of time.
The solvent has to be non-toxic, because a residue of it will always remain in the powder mixture.
The active substance has to be stable in solution.
For practical examples of this method see Sect. 4.6.2.
4.5.2 Colouring of Powder Mixtures
Capsule contents can be coloured by using a coloured powder mixture as diluent. Powders can be coloured with water-soluble, or water insoluble colouring agents (see Table 23.26). Water-soluble colouring agents should be dissolved in order to produce an even distribution over the diluent. The use of water to promote the dispersion of the water-soluble colouring agent may cause granulation of particles. To prevent or minimise this event the addition of the solution to the powder must be slowly and with continuous stirring. Water is a safe solvent for everyone, but evaporates slowly. Ethanol in a concentration of more than 90 % evaporates quickly, but the use of organic solvents has to be considered carefully because of the possible residues. Water-insoluble colouring agents, on the other hand, mix well with the diluent in the dry state.
4.6 Capsules
Capsules are probably the most versatile dosage form prepared in the pharmacy. Capsules may contain one active substance or a mixture of active substances, usually a diluent and sometimes a glidant or a disintegrating agent or both. Capsules are easy to swallow and can enclose active substances with an unpleasant taste. Capsules can be prepared in the range of few units up to hundreds of thousand units. Further advantages, namely by comparison to tablets, are the small number of excipients required to prepare capsules, and the possibility of having non-compressed powders, allowing a faster dissolution of the active substance.
4.6.1 Capsule Shells
Capsules consist of a shell made of a structural polymer (e.g. gelatine, hypromellose, starch) together with other excipients that allow the preparation of the shell itself (e.g. plasticizers) or provide some functionality to the shell (e.g. titanium oxide for making capsules opaque). Gelatine is often extracted from animals (e.g. pig), that is why some people doesn’t want to ingest them. The pharmacist must consider possible interactions between the active substance or the excipients and gelatine. Alternatively, ‘vegetarian’ capsules exist, which consist of gelatine from algae or of a cellulose derivative.
Empty capsule shells are stored at room temperature in tight containers that maintain a constant, adequate relative humidity. The Deutsche Arzneimittel Codex (DAC) describes the test for dissolution of capsule shells: empty capsules should dissolve or at least open in less than 15 min [30]. Incorrectly stored capsules do not dissolve, they just swell. Furthermore cycles of high and low humidity and temperature damage the shells irreversibly.
In pharmacies hard capsule shells consisting of a body and a cap with a locking mechanism are by far the most used. These shells come in various sizes: size 5 being the smallest and size 00 the largest. Many patients experience difficulty in swallowing large capsules. Usually capsule sizes 1 or 2 for adults and size 3 or 4 for children are used in pharmacy preparation. In some cases the patient is instructed to open the capsules to take the contents with food or dissolve it in water, provided that capsules can be opened and the drug can be given without the protection of the capsule shell.
4.6.2 Different Methods for Preparing the Powder Mass
As an addition to the general method of preparation of a powder mixture, for capsules some specific directions apply. Capsules are filled by volume, therefore, the powder mixture should be prepared to obtain a specific volume. The correct volume depends on the capsule size and the number of capsules to be filled (Table 4.3).
Table 4.3
Volumes of microcrystalline cellulose filled in hard gelatine capsule shells (in cm3)
Number | Capsule size | ||||
|---|---|---|---|---|---|
00 | 0 | 1 | 2 | 3 | |
20 | 18 | 13 | |||
30 | 28 | 20 | 14 | 11 | |
50 | 46 | 34 | 24 | 19 | 14 |
60 | 55 | 40 | 29
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

| ||