Opioids, Analgesia, and Pain Management
Pain is a component of virtually all clinical pathologies, and management of pain is a primary clinical imperative. Opioids are a mainstay of pain treatment, but depending upon the pain state, therapy may involve NSAIDs, anticonvulsants, or antidepressants.
The term opiate refers to compounds structurally related to products found in opium derived from the resin of the opium poppy, Papaver somniferum. Opiates include the natural plant alkaloids, such as morphine, codeine, thebaine, and many semisynthetic derivatives. An opioid is any agent that has the functional and pharmacological properties of an opiate. Endogenous opioids are naturally occurring ligands for opioid receptors found in animals. The term endorphin is used synonymously with endogenous opioid peptides but also refers to a specific endogenous opioid, β-endorphin. Although the term narcotic originally referred to any drug that induced narcosis or sleep, the word has become associated with opioids and is often used in a legal context to refer to substances with abuse or addictive potential.
ENDOGENOUS OPIOID PEPTIDES
Several distinct families of endogenous opioids peptides have been identified: principally the enkephalins, endorphins, and dynorphins (Table 18–1). These families have several common properties:
Table 18–2
Endogenous Opioid Peptides
• Each derives from a distinct precursor protein, pre-pro-opiomelanocortin (pre-POMC), preproenkephalin, and preprodynorphin, respectively, encoded by a corresponding gene.
• Each precursor is subject to complex cleavages by distinct trypsin-like enzymes, and to a variety of posttranslational modifications resulting in the synthesis of multiple peptides, some of which are active.
• Most opioid peptides with activity at a receptor share the common amino-terminal sequence of Tyr-Gly-Gly-Phe-(Met or Leu) followed by various C-terminal extensions yielding peptides of 5-31 residues; the endomorphins, with different terminal sequences, are exceptions.
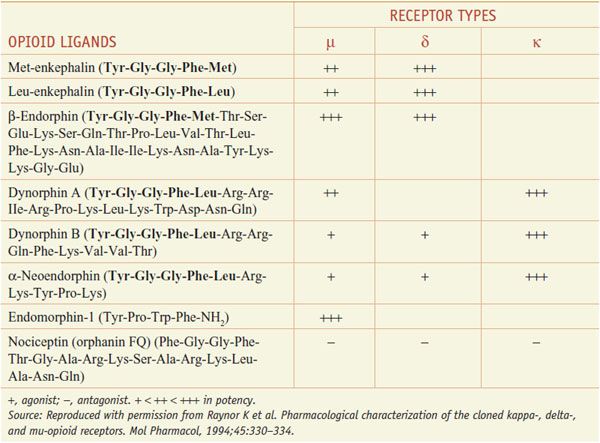
The opioid peptide precursors are a protean family (Figure 18–1). The major opioid peptide derived from POMC is the potent opioid agonist, β-endorphin. The POMC sequence also is processed into a variety of nonopioid peptides including adrenocorticotropic hormone (ACTH), melanocyte-stimulating hormone (α-MSH), and β-lipotropin (β-LPH). Although β-endorphin contains the sequence for met-enkephalin at its amino terminus, it is not converted to this peptide. Proenkephalin contains multiple copies of met-enkephalin, as well as a single copy of leu-enkephalin. Prodynorphin contains 3 peptides of differing lengths that all begin with the leu-enkephalin sequence: dynorphin A, dynorphin B, and neoendorphin. Nociceptin peptide or orphanin FQ (now termed N/OFQ) shares structural similarity with dynorphin A.
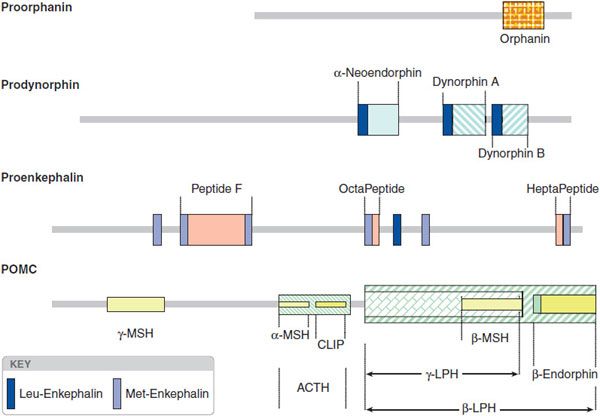
Figure 18–1 Peptide precursors. (Reproduced with permission from Akil H, Owens C, Gustein H, et al. Endogenous opioids: Overview and current issues. Drug Alcohol Depend, 1998;51:127-140. Copyright © Elsevier.)
Endomorphins belong to a novel family of peptides that include: endomorphin-1 (Tyr-Pro-Trp-Phe-NH2) and endomorphin-2 (Tyr-Pro-Phe-Phe-NH2). Endomorphins have an atypical structure and display selectivity towards the μ opioid receptor. Several points should be stressed:
• Not all cells that make a given opioid prohormone precursor store and release the same mixture of opioid peptides; this results from differential processing secondary to variations in the cellular complement of peptidases that produce and degrade the active opioid fragments.
• Processing of these peptides is altered by physiological demands, leading to the release of a different mix of posttranslationally derived peptides by a given cell under different conditions.
• Opioid peptides are found in plasma and reflect release from secretory systems such as the pituitary and the adrenals that do not reflect neuraxial release. Conversely, levels of these peptides in brain/spinal cord and in cerebrospinal fluid (CSF) arise from neuraxial systems and not from peripheral systems.
OPIOID RECEPTORS
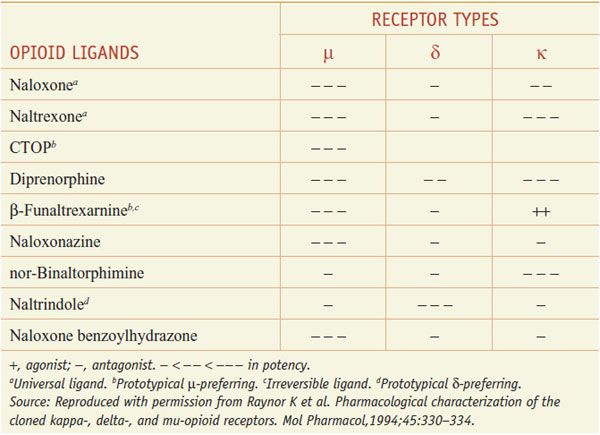
The 3 opioid receptors—μ, δ, and κ (MOR, DOR, and KOR)—belong to the rhodopsin family of GPCRs (see Chapter 3) and share extensive sequence homologies (55-58%). Highly selective agonists have been developed that show specific affinity for the respective binding site (e.g., DAMGO for μ, DPDPE for δ, and U-50,488, and U-69,593 for κ) (Table 18–2). Commonly used antagonists (Table 18–3) include cyclic analogs of somatostatin such as CTOP as a μ receptor antagonist, a derivative of naloxone called naltrindole as a δ receptor antagonist, and a bivalent derivative of naltrexone called nor-binaltorphimine (nor-BNI) as a κ-receptor antagonist.
Table 18–2
Opioid Agonists
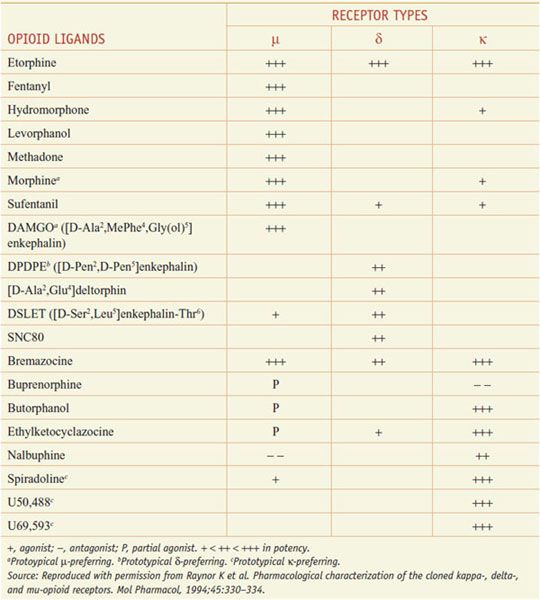
Table 18–3
Opioid Antagonists
In the membrane, opiate receptors can form both homo- and heterodimers. Dimerization can alter the pharmacological properties of the respective receptors. Splice variants exist within each of the 3 opioid receptor families, and this alternative splicing of receptor transcripts may be crucial for the diversity of opioid receptors. Given the functional importance of the intracellular components of the GPCRs, it is not surprising that significant differences exist for the receptor isoforms in terms of agonist-induced G protein activation and receptor internalization.
An opiate receptor-like protein (ORL1 or NOP) was cloned based on its structural homology (48-49% identity) to other members of the opioid receptor family; it is G-protein coupled, has an endogenous ligand (nociception/orphanin, FQ: N/OFQ), but does not display an opioid pharmacology.
OPIATE RECEPTOR SIGNALING
The μ, δ, and κ receptors couple through pertussis toxin-sensitive, Gi/Go proteins (but occasionally to Gs or Gz). Upon receptor activation, the Gi/Go coupling results in a large number of intracellular events, including:
• Inhibition of adenylyl cyclase activity
• Reduced opening of voltage-gated Ca2+ channels (reduces neurotransmitter release from presynaptic terminals)
• Stimulation of K+ current through several channels including G protein-activated inwardly rectifying K+ channels (GIRKs) (hyperpolarizes and inhibits postsynaptic neurons)
• Activation of PKC and PLCβ
FUNCTIONAL CONSEQUENCES OF ACUTE AND CHRONIC OPIATE RECEPTOR ACTIVATION
The loss of effect with exposure to opiates occurs over short- and long-term intervals.
INTERNALIZATION. μ and δ receptors can undergo rapid agonist-mediated internalization via a classic endocytic, β-arrestin-mediated pathway, whereas κ receptors do not internalize after prolonged agonist exposure. Internalization of the μ and δ receptors may be induced differentially as a function of the structure of the ligand.
DESENSITIZATION. In the face of a transient activation (minutes to hours), acute tolerance or desensitization occurs that is specific for that receptor and disappears with a time course parallel to the clearance of the agonist. Short-term desensitization probably involves phosphorylation of the receptors resulting in an uncoupling of the receptor from its G-protein and/or internalization of the receptor.
TOLERANCE. Here tolerance refers to a decrease in the apparent effectiveness of a drug with continuous or repeated agonist administration (days to weeks), that, following removal of the agonist, disappears over several weeks. This tolerance is reflected by a reduction in the maximum achievable effect or a right shift in the dose-effect curve. This loss of effect with persistent exposure to an opiate agonist has several key properties:
• Different physiological responses develop tolerance at different rates. Thus, at the organ system level, some end points show little or no tolerance development (pupillary miosis), some show moderate tolerance (constipation, emesis, analgesia, sedation), and some show rapid tolerance (euphorigenic).
• In general, opiate agonists of a given class will typically show a reduced response in a system rendered tolerant to another agent of that class (e.g., cross-tolerance between the μ agonists, such as morphine and fentanyl). This cross-tolerance is not consistent or complete, thereby forming the basis for the switching between opioid drugs in clinical therapy.
DEPENDENCE. Dependence represents a state of adaptation manifested by receptor/drug class-specific withdrawal syndrome produced by cessation of drug exposure (e.g., by drug abstinence) or administration of an antagonist (e.g., naloxone). At the organ system level, withdrawal is manifested by significant somatomotor and autonomic outflow (reflected by agitation, hyperalgesia, hyperthermia, hypertension, diarrhea, pupillary dilation, and release of virtually all pituitary and adrenomedullary hormones) and by affective symptoms (dysphoria, anxiety, and depression).
ADDICTION. Addiction is a behavioral pattern characterized by compulsive use of a drug. The positive, rewarding effects of opiates are considered to be the driving component for initiating the recreational use of opiates. This positive reward property is subject to the development of tolerance. Given the aversive nature of withdrawal symptoms, avoidance and alleviation of withdrawal symptoms may become a primary motivation for compulsive drug taking. Drug dependence is not synonymous with drug addiction. Tolerance and dependence are physiological responses seen in all patients but are not predictors of addiction (see Chapter 24). For example, cancer pain often requires prolonged treatment with high doses of opioids, leading to tolerance and dependence. Yet abuse in this setting is considered to be unusual.
MECHANISMS OF TOLERANCE/DEPENDENCE-WITHDRAWAL. The mechanisms underlying chronic tolerance and dependence/withdrawal are controversial. Several types of events may to contribute.
Receptor Disposition. Acute desensitization or receptor internalization may play a role in the initiation of chronic tolerance but is not sufficient to explain persistent changes observed with chronic exposure. Thus, morphine, unlike other μ agonists, does not promote μ receptor internalization or receptor phosphorylation and desensitization. Receptor desensitization and downregulation are agonist specific. Endocytosis and sequestration of receptors do not invariably lead to receptor degradation but can also result in receptor dephosphorylation and recycling to the surface of the cell. Accordingly, opioid tolerance may not be related to receptor desensitization but rather to a lack of desensitization. Agonists that rapidly internalize opioid receptors also would rapidly desensitize signaling, but this desensitization would be at least partially reset by recycling of “reactivated” opioid receptors.
Adaptation of Intracellular Signaling Mechanisms in the Opioid Receptor-Bearing Neurons. Coupling of MOR to cellular effectors, such as inhibition of adenylyl cyclase, activation of inwardly rectifying K+ channels, inhibition of Ca2+ currents, and inhibition of terminal release of transmitters demonstrates functional uncoupling of receptor occupancy from effector function. Importantly, the chronic opioid effect initiates adaptive counter-regulatory change. The best example of such cellular counter-regulatory processes is the rebound increase in cellular cyclic AMP levels produced by “superactivation” of adenylyl cyclase and upregulation of the amount of enzyme.
System Level Counter-Adaptation. With chronic opiate exposure, there is a loss of drug effect; this may reflect an enhanced excitability of the regulated link. Thus, tolerance to the analgesic action of chronically administered μ opiates may result in an activation of bulbospinal pathways that increases the excitability of spinal dorsal horn pain transmission linkages. With chronic opiate exposure, opiate receptor occupancy will lead to the activation of PKC, which can phosphorylate and, accordingly, enhance the activation of local NMDA glutamate receptors (see Chapter 14). These receptors mediate a facilitated state leading to enhanced spinal pain processing. Blocking of these receptors can at least partially attenuate the loss of analgesic efficacy with continued opiate exposure. These system level counter-adaptation hypotheses represent mechanisms that may apply to specific systems (e.g., pain modulation) but not necessarily to others (e.g., sedation or miosis).
EFFECTS OF CLINICALLY USED OPIOIDS
Opiates, depending on their receptor specificities, produce a variety of effects consistent with the role played by the organ systems with which the receptors are associated. Although the primary clinical use of opioids is for their pain-relieving properties, opioids produce a host of other effects. This is not surprising in view of the wide distribution of opioid receptors in the brain and the periphery.
ANALGESIA. Morphine-like drugs produce analgesia, drowsiness, and euphoria (changes in mood, and mental clouding). When therapeutic doses of morphine are given to patients with pain, they report the pain to be less intense or entirely gone. In addition to relief of distress, some patients may experience euphoria. Analgesia often occurs without loss of consciousness, though drowsiness commonly occurs. Morphine at these doses does not have anticonvulsant activity and usually does not cause slurred speech, emotional lability, or significant motor uncoordination. When morphine in an analgesic dose is given to normal, pain-free individuals, the patients may report the drug experience to be frankly unpleasant. There may be drowsiness, difficulty in mentation, apathy, and lessened physical activity. As the dose is increased, the subjective, analgesic, and toxic effects, including respiratory depression, become more pronounced. The relief of pain by morphine-like opioids is selective in that other sensory modalities, such as light touch, proprioception, and the sense of moderate temperatures, are unaffected. Low doses of morphine produce reductions in the affective but not the perceived intensity of pain. Continuous dull pain (as generated by tissue injury and inflammation) is relieved more effectively than sharp intermittent (incident) pain, such as that associated with the movement of an inflamed joint, but with sufficient amounts of opioid it is possible to relieve even the severe piercing pain associated with acute renal or biliary colic.
PAIN STATES AND MECHANISMS UNDERLYING DIFFERENT PAIN STATES
Acute Nociception. Acute activation of small high-threshold sensory afferents (Aδ and C fibers) generates transient input into the spinal cord, which in turn leads to activation of neurons that project contralaterally to the thalamus and thence to the somatosensory cortex. A parallel spinofugal projection is to the medial thalamus and from there to the anterior cingulate cortex, part of the limbic system. The output produced by acutely activating this ascending system is sufficient to evoke pain reports. Examples of such stimuli include a hot coffee cup, a needle stick, or an incision.
Tissue Injury. Following tissue injury or local inflammation (e.g., local skin burn, toothache, rheumatoid joint), an ongoing pain state arises that is characterized by burning, throbbing, or aching and an abnormal pain response (hyperalgesia) and can be evoked by otherwise innocuous or mildly aversive stimuli (tepid bathwater on a sunburn; moderate extension of an injured joint). This pain typically reflects the effects of active factors (such as prostaglandins, bradykinin, cytokines, and H+ ions, among many mediators) released into the injury site, which have the ability to activate the terminal of small high-threshold afferents (Aδ and C fibers) and to reduce the stimulus intensity required to activate these sensory afferents (peripheral sensitization). In addition, the ongoing afferent traffic initiated by the injury leads to the activation of spinal facilitatory cascades, yielding a greater output to the brain for any given input. This facilitation is thought to underlie the hyperalgesic states. Such tissue injury-evoked pain is often referred to as “nociceptive” pain (Figure 18–2). Examples of such states would be burn, post-incision, abrasion of the skin, inflammation of the joint, and musculoskeletal injury.
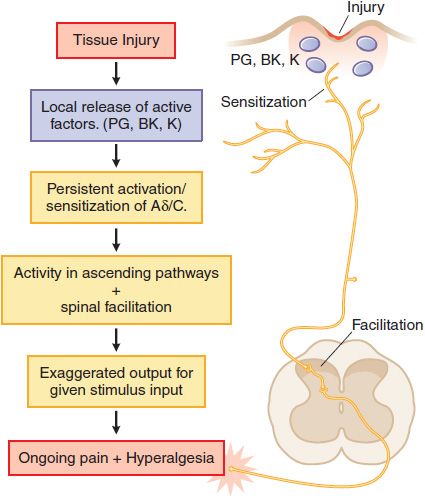
Figure 18–2 Mechanisms of tissue injury-evoked nociception.
Nerve Injury. Injury to the peripheral nerve yields complex anatomical and biochemical changes in the nerve and spinal cord that induce spontaneous dysesthesias (shooting, burning pain) and allodynia (light touch hurts). This nerve injury pain state may not depend on the activation of small afferents, but may be initiated by low-threshold sensory afferents (e.g., Aβ fibers). Such nerve injuries result in the development of ectopic activity arising from neuromas formed by nerve injury and the dorsal root ganglia of the injured axons as well as a dorsal horn reorganization, such that low-threshold afferent input carried by Aβ fibers evokes a pain state. Examples of such nerve injury-inducing events include nerve trauma or compression (carpal tunnel syndrome), chemotherapy (as for cancer), diabetes, and in the post-herpetic state (shingles). These pain states are said to be neuropathic (Figure 18–3). Many clinical pain syndromes, such as found in cancer, typically represent a combination of these inflammatory and neuropathic mechanisms. Although nociceptive pain usually is responsive to opioid analgesics, neuropathic pain is typically considered to respond less well to opioid analgesics.
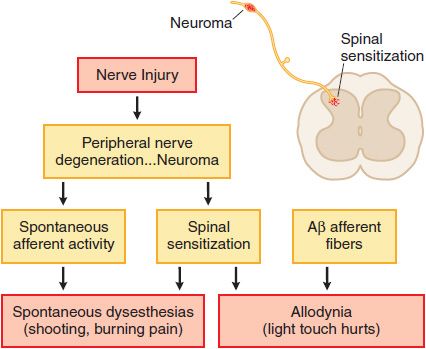
Figure 18–3 Mechanisms of nerve injury-evoked nociception.
Sensory Versus Affective Dimensions. When pain does not evoke its usual responses (anxiety, fear, panic, and suffering), a patient’s ability to tolerate the pain may be markedly increased, even when the capacity to perceive the sensation is relatively unaltered. It is clear, however, that alteration of the emotional reaction to painful stimuli is not the sole mechanism of analgesia. Thus, intrathecal administration of opioids can produce profound segmental analgesia without causing significant alteration of motor or sensory function or subjective effects.
MECHANISMS OF OPIOID-INDUCED ANALGESIA. The analgesic actions of opiates after systemic delivery represent actions in the brain, spinal cord, and in some instances in the periphery.
Supraspinal Actions. Microinjections of morphine into the mesencephalic periaqueductal gray (PAG) matter region will block nociceptive responses; naloxone will reverse these effects. Several mechanisms exist whereby opiates with an action limited to the PAG may act to alter nociceptive transmission. These are summarized in Figure 18–4. MOR agonists block release of the inhibitory transmitter GABA from tonically active PAG systems that regulate activity in projections to the medulla. PAG projections to the medulla activate medullospinal release of NE and 5HT at the level of the spinal dorsal horn. This release can attenuate dorsal horn excitability. Interestingly, this PAG organization can also serve to increase excitability of dorsal raphe and locus coeruleus from which ascending serotonergic and noradrenergic projections to the limbic forebrain originate.
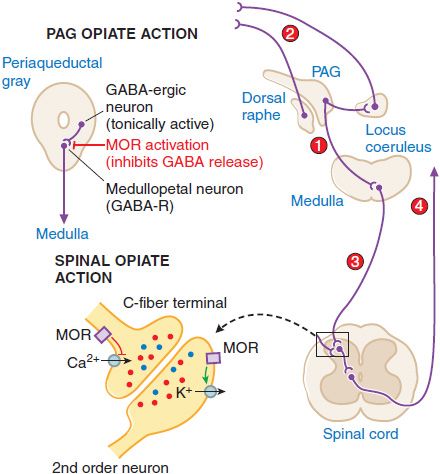
Figure 18–4 Mechanisms of opiate action in producing analgesia. Top left: Schematic of organization of opiate action in the periaqueductal gray. Top right: Opiate-sensitive pathways in PAG μ-opiate actions block the release of GABA from tonically active systems that other-wise regulate the projections to the medulla (1) leading to an activation of PAG outflow resulting in activation of forebrain (2) and spinal (3) monoamine receptors that regulate spinal cord projections (4), which provide sensory input to higher centers and mood. Bottom left: Schematic of primary afferent synapse with second-order dorsal horn spinal neuron, showing pre- and postsynaptic opiate receptors coupled to Ca2+ and K+ channels, respectively. Opiate-receptor binding is highly expressed in the superficial spinal dorsal horn (substantia gelatinosa). These receptors are located pre-synaptically on the terminals of small primary afferents (C fibers) and postsynaptically on second-order neurons. Presynaptically, activation of MOR blocks the opening of the voltage-sensitive Ca2+ channel, which otherwise initiates transmitter release. Postsynaptically, MOR activation enhances opening of K+ channels, leading to hyperpolarization. Thus, an opiate agonist acting at these sites jointly serves to attenuate the afferent-evoked excitation of the second-order neuron.
Spinal Opiate Action. A local action of opiates in the spinal cord will selectively depress the discharge of spinal dorsal horn neurons evoked by small (high-threshold) but not large (low-threshold) afferent nerve fibers. Intrathecal administration of opioids in animals ranging from mouse to human will reliably attenuate the response of the organism to a variety of somatic and visceral stimuli that otherwise evoke pain states. Specific opiate binding and receptor protein are limited for the most part to the substantia gelatinosa of the superficial dorsal horn, the region in which small, high-threshold sensory afferents show their principal termination. A significant proportion of these opiate receptors are associated with small peptidergic primary afferent C fibers; the remainder are on local dorsal horn neurons.
Spinal opiates reduce the release of primary afferent peptide transmitters such as substance P contained in small afferents. The presynaptic action corresponds to the ability of opiates to prevent the opening of voltage-sensitive Ca+ channels, thereby preventing transmitter release. A postsynaptic action is demonstrated by the ability of opiates to block excitation of dorsal horn neurons directly evoked by glutamate, reflecting a direct activation of dorsal horn projection neurons. The activation of K+ channels in these postsynaptic neurons, leading to hyperpolarization, is consistent with a direct postsynaptic inhibition. The joint capacity of spinal opiates to reduce the release of excitatory neurotransmitters from C fibers and to decrease the excitability of dorsal horn neurons is believed to account for the powerful and selective effect of opiates on spinal nociceptive processing. A variety of opiates delivered spinally (intrathecally or epidurally) can induce a powerful analgesia that is reversed by low doses of systemic naloxone.
Peripheral Action. Direct application of opiates to a peripheral nerve can, in fact, produce a local anesthetic-like action at high concentrations, but this is not naloxone reversible and is believed to reflect a “nonspecific” action. Conversely, direct injection of these agents into peripheral sites have demonstrated that under conditions of inflammation, where there is an increased terminal sensitivity leading to an exaggerated pain response (e.g., hyperalgesia), the local action of opiates can exert a normalizing effect on the exaggerated thresholds. Whether the effects are uniquely on the afferent terminal, whether the opiate acts on inflammatory cells that release products that sensitize the nerve terminal, or both, is not known.
MOOD ALTERATIONS AND REWARDING PROPERTIES. The mechanisms by which opioids produce euphoria, tranquility, and other alterations of mood (including rewarding properties) are complex and not entirely clear. Neural systems that mediate opioid reinforcement overlap with, but are distinct from, those involved in physical dependence and analgesia. Behavioral and pharmacological data point to a pivotal role of the mesocorticolimbic dopamine system that projects to the nucleus accumbens (NAc) in drug-induced reward and motivation (Figure 18–5).
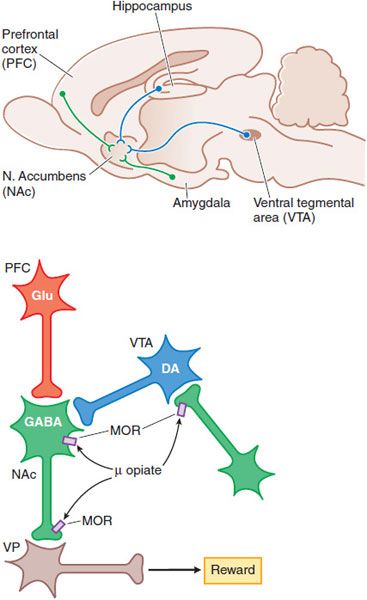
Figure 18–5 Schematic pathways underlying rewarding properties of opiates. Upper panel: This sagittal section of rat brain shows DA and GABA inputs from the ventral tegmental area (VTA) and prefrontal cortex (PFC), respectively, into the nucleus accumbens (NAc). Lower panel: Neurons are labeled with their primary neurotransmitters. At a cellular level, MOR agonists reduce excitability and transmitter release at the sites indicated by inhibiting Ca2+ influx and enhancing K+ current (see Figure 18–4). Thus, opiate-induced inhibition in the VTA on GABA-ergic interneurons or in the NAc reduce GABA-mediated inhibition and increase outflow from the ventral pallidum (VP), which appears to correlate with a positive reinforcing state (enhanced reward).
RESPIRATION. Although effects on respiration are readily demonstrated, clinically significant respiratory depression rarely occurs with standard analgesic doses in the absence of other contributing variables (discussed in the next sections). It should be stressed, however, that respiratory depression represents the primary cause of morbidity secondary to opiate therapy. In humans, death from opiate poisoning is nearly always due to respiratory arrest or obstruction. Opiates depress all phases of respiratory activity (rate, minute volume, and tidal exchange) and produce irregular and aperiodic breathing. The diminished respiratory volume is due primarily to a slower rate of breathing; with toxic amounts of opioids, the rate may fall to 3-4 breaths/min. Thus, opioids must be used with caution in patients with asthma, chronic obstructive pulmonary disease (COPD), cor pulmonale, decreased respiratory reserve, preexisting respiratory depression, hypoxia, or hypercapnia to avoid apnea due to a decrease in respiratory drive coinciding with an increased airway resistance. Although respiratory depression is not considered to be a favorable therapeutic effect of opiates, their ability to suppress respiratory drive is used to therapeutic advantage to treat dyspnea resulting, e.g., in patients with COPD, where air hunger leads to extreme agitation, discomfort, and gasping; similarly, opiates find use in patients who require artificial ventilation.
Morphine-like opioids depress respiration through μ receptors and δ receptors in part by a direct depressant effect on rhythm generation. A key property of opiate effects on respiration is the depression of the ventilatory response to increased CO2. Opiates also will depress ventilation otherwise driven by hypoxia through an effect on carotid and aortic body chemosensors. Importantly, with opiates, hypoxic stimulation of chemoreceptors still may be effective when opioids have decreased the responsiveness to CO2, and inhalation of O2 may remove the residual drive resulting from the elevated PO2 and produce apnea. In addition to the effect upon respiratory rhythm and chemosensitivity, opiates can have mechanical effects on airway function by increasing chest wall rigidity and diminishing upper airway patency.
FACTORS EXACERBATING OPIATE-INDUCED RESPIRATORY DEPRESSION. A number of factors are recognized as increasing the risk of opiate-related respiratory depression even at therapeutic doses:
• Other medications. The combination of opiates with other depressant medications, such as general anesthetics, tranquilizers, alcohol, or sedative-hypnotics, produces additive depression of respiratory activity.
• Sleep. Natural sleep decreases the sensitivity of the medullary center to CO2, and the depressant effects of morphine and sleep are at least additive. Obstructive sleep apnea is considered to be an important risk factor for increasing the likelihood of fatal respiratory depression.
• Age. Newborns can show significant respiratory depression and desaturation; and this may be evident in lower Apgar scores if opioids are administered parenterally to women within 2-4 h of delivery because of transplacental passage of opioids. Elderly patients are at greater risk of depression because of reduced lung elasticity, chest wall stiffening, and decreased vital capacity.
• Disease. Opiates may cause a greater depressant action in patients with chronic cardiopulmonary or renal diseases because they can manifest a desensitization of the response to increased CO2.
• COPD. Enhanced depression can also be noted in patients with COPD and sleep apnea secondary to diminished hypoxic drive (although morphine can relieve gasping in late-stage COPD by this mechanism).
• Relief of Pain. Pain stimulates respiration; removal of the painful condition (as with the analgesia resulting from the therapeutic use of the opiate) will reduce the ventilatory drive and lead to apparent respiratory depression.
Respiratory depression produced by any opiate agonist can be readily reversed by delivery of an opiate antagonist. Opiate antagonist reversal in the somnolent patient is considered to be indicative of an opiate-mediated somnolence. It is important to remember that most opiate antagonists have a relatively short duration of action as compared to an agonist such as morphine or methadone and fatal “re-narcotization” can occur if vigilance is not exercised.
NEUROENDOCRINE EFFECTS. The regulation of the release of hormones and factors from the pituitary is under complex regulation by opiate receptors in the hypothalamic-pituitary-adrenal (HPA) axis. Broadly considered, morphine-like opioids block the release of a large number of HPA hormones.
Sex Hormones. In males, acute opiate therapy reduces plasma cortisol, testosterone, and gonadotrophins. Inhibition of adrenal function is reflected by reduced cortisol production and reduced adrenal androgens (dehydroepiandrosterone, DHEA). In females, morphine will additionally result in lower LH and FSH release. In both males and females, chronic therapy can result in endocrinopathies, including hypogonadotrophic hypogonadism. In men, this may result in decreased libido and, with extended exposure, reduced secondary sex characteristics. In women these exposures are associated with menstrual cycle irregularities. These changes are reversible with removal of the opiate.
Prolactin. Prolactin release from the anterior pituitary is under inhibitory control by DA released from neurons of the arcuate nucleus. MOR agonists act presynaptically on these DA-releasing terminals to inhibit DA release and thereby increase plasma prolactin.
Antidiuretic Hormone and Oxytocin. KOR agonists inhibit the release of oxytocin and antidiuretic hormone (and cause prominent diuresis). It should be noted that agents such as morphine may yield a hypotension secondary to histamine release and this would, by itself, promote ADH release.
MIOSIS. MOR agonists induce pupillary constriction (miosis) in the awake state and block pupillary reflex dilation during anesthesia. The parasympathetic outflow is locally regulated by GABA-ergic interneurons. Opiates are believed to block the GABA-ergic interneuron-mediated inhibition.
OTHER EFFECTS
SEIZURES AND CONVULSIONS. In older children and adults, moderately higher doses of opiates produce EEG slowing. In the newborn, morphine has been shown to produce epileptiform activity and occasionally seizure activity. Several mechanisms are most certainly involved in these excitatory actions:
• Inhibition of inhibitory interneurons. Morphine-like drugs excite certain groups of neurons, especially hippocampal pyramidal cells, probably from inhibition of the release of GABA by interneurons.
• Direct stimulatory effects.
• Actions mediated by non-opioid receptors. The metabolites of several opiates (morphine-3-glucuronide, normeperidine) have been implicated in seizure activity.
COUGH. Morphine and related opioids depress the cough reflex at least in part by a direct effect on a cough center in the medulla and this can be achieved without altering the protective glottal function. Cough is a protective reflex evoked by airway stimulation. It involves rapid expression of air against a transiently closed glottis.
NAUSEANT AND EMETIC EFFECTS. Nausea and vomiting produced by morphine-like drugs are side effects caused by direct stimulation of the chemoreceptor trigger zone for emesis in the area postrema of the medulla.
CARDIOVASCULAR SYSTEM. In the supine patient, therapeutic doses of morphine-like opioids have no major effect on blood pressure or cardiac rate and rhythm. Such doses can, however, produce peripheral vasodilation, reduced peripheral resistance, and an inhibition of baroreceptor reflexes. Thus, when supine patients assume the head-up position, orthostatic hypotension and fainting may occur. The peripheral arteriolar and venous dilation produced by morphine involves several mechanisms:
• Morphine induces release of histamine from mast cells, leading to vasodilation; this effect is reversed by naloxone but only partially blocked by H1 antagonists
• Morphine blunts reflex vasoconstriction caused by increased PCO2
Morphine may exert its therapeutic effect in the treatment of angina pectoris and acute myocardial infarction by decreasing preload, inotropy, and chronotropy, thus favorably altering determinants of myocardial O2 consumption. Morphine has been shown to produce cardioprotective effects. Morphine can mimic the phenomenon of ischemic preconditioning, where a short ischemic episode paradoxically protects the heart against further ischemia. This effect appears to be mediated through receptors signaling through a mitochondrial ATP-sensitive K+ channel in cardiac myocytes; the effect also is produced by other GPCRs signaling through Gi. Morphine-like opioids should be used with caution in patients who have decreased blood volume because these agents can aggravate hypovolemic shock. Morphine should be used with great care in patients with cor pulmonale; deaths after ordinary therapeutic doses have been reported. Concurrent use of certain phenothiazines may increase the risk of morphine-induced hypotension.
MOTOR TONE. High doses of opioids, as used for anesthetic induction, produce muscular rigidity. Myoclonus, ranging from mild twitching to generalized spasm, is an occasional side effect, which has been reported with all clinical opiate agonists; it is particularly prevalent in patients receiving high doses. Increase motor tone and rigidity are reversed by opiate antagonists.
GI TRACT. Between 40% and 95% of patients treated with opioids develop constipation and changes in bowel function. Opioid receptors are densely distributed in enteric neurons between the myenteric and submucosal plexi and on a variety of secretory cells.
Esophagus. Morphine inhibits lower esophageal sphincter relaxation induced by swallowing and by esophageal distension; the effect is believed to be centrally mediated.
Stomach. Morphine increases in tonic contracture of the antral musculature and upper duodenum and reduces resting tone in the musculature of the gastric reservoir, thereby prolonging gastric emptying time and increasing the likelihood of esophageal reflux. Passage of the gastric contents through the duodenum may be delayed by as much as 12 h, and the absorption of orally administered drugs is retarded. Morphine and other agonists usually decrease secretion of hydrochloric acid.
Intestine.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


