Nutrition in Liver Disorders and the Role of Alcohol1
Juliane I. Beier
Sarah Landes
Mohammad Mohammad
Craig J. Mcclain
1Abbreviations: ADH, alcohol dehydrogenase; AH, alcoholic hepatitis; ALD, alcoholic liver disease; ALDH, aldehyde dehydrogenase; BAC, blood alcohol concentration; BCAA, branched-chain amino acid; CNS, central nervous system; DPI, diphenyliodonium; FAEE, fatty acid ethyl ester; GSH, glutathione; HE, hepatic encephalopathy; HIF, hypoxia-inducible factor; iNOS, inducible nitric oxide synthase; LES, late evening snack; LPS, lipopolysaccharide; NAD+, oxidized nicotinamide adenine dinucleotide; NADH, reduced nicotinamide adenine dinucleotide; NADP+, oxidized nicotinamide adenine dinucleotide phosphate; NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis; PEM, protein-energy malnutrition; RNS, reactive nitrogen species; ROS, reactive oxygen species; RQ, respiratory quotient; SAM, S-adenosylmethionine; SEC, sinusoidal endothelial cell; TLR-4, Toll-like receptor-4; TNF, tumor necrosis factor; TRIF, TIR-domain-containing adapter-inducing interferon-β; VA, Veterans Affairs; WKS, Wernicke-Korsakoff syndrome.
OVERVIEW OF THE LIVER AND ALCOHOL METABOLISM
The liver is the largest organ in the body, and it has a unique dual blood supply, perfused by both the portal vein (directly exposed to absorbed nutrients) and the hepatic artery. The liver is composed of multiple cell types with differing functions. Hepatocytes make up more than 80% of total liver mass and play a critical role in the metabolism of amino acids and ammonia, lipids, carbohydrates, vitamins, minerals, hormones, and detoxification of a variety of drugs and xenobiotics. Hepatic stellate cells comprise the major storehouse for vitamin A in the body, and they play a critical role in collagen formation during liver injury and fibrosis. Sinusoidal endothelial cells (SECs) make up approximately half of the nonparenchymal cells of the liver and play an important role in controlling the exchange of materials (including nutrients) between the bloodstream and the liver parenchyma. The SECs express scavenger receptors and act as antigen-presenting cells, to name only a few of the important immune functions of these cells. Hepatic Kupffer cells comprise the largest reservoir of fixed macrophages in the body. They play a protective role against gut-derived toxins that have escaped into the portal circulation, and they are a major producer of cytokines, which can markedly influence nutritional status. Bile duct epithelial cells play a major role in transport (e.g., water, bile), express a variety of transporters, and have important immune functions. All these cell types interact in a coordinated fashion to protect against gutderived toxins and autoimmune reactions (tolerance), and they modulate hormonal and nutritional status.
The liver is also the main organ for ethanol metabolism. Ethanol is primarily metabolized (˜80%) by alcohol dehydrogenase (ADH), oxidizing ethanol to acetaldehyde. However, cytochrome P-450 systems (mainly CYP2E1) and catalase also contribute to the production of acetaldehyde in ethanol metabolism. Whereas ADH transfers electrons from ethanol to the reducing equivalent oxidized nicotinamide adenine dinucleotide phosphate (NADP+), the cytochrome P-450 system transfers the electrons to molecular oxygen (O2), and catalase reduces hydrogen peroxide (H2O2) to water (Fig. 82.1). Ethanol metabolism is considered zero order at physiologically relevant doses, meaning that the oxidation of ethanol is saturated at blood alcohol concentrations (BACs) that cause significant central nervous system (CNS) effects (>0.03%). Whereas the oxidation of ethanol to acetaldehyde is mediated by three distinct enzyme systems, only one enzyme, aldehyde dehydrogenase (ALDH), oxidizes acetaldehyde to acetate (see Fig. 82.1). Analogous to ADH, ALDH uses oxidized nicotinamide adenine dinucleotide (NAD+) as the electron acceptor for this reaction; however, ALDH is located in the mitochondria of the cell (see Fig. 82.1).
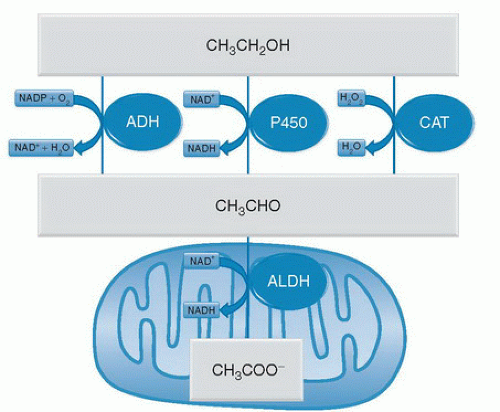 Fig. 82.1. Oxidative metabolism of alcohol by the liver. Alcohol (CH3CH2OH) is oxidized to acetaldehyde (CH3CHO) by three enzyme systems: the microsomal ethanol oxidizing system (MEOS), alcohol dehydrogenase (ADH), and catalase (CAT). Acetaldehyde, in turn, is metabolized to acetate (CH3COO-) by aldehyde dehydrogenase (ALDH) in the mitochondria. The metabolic and biochemical effects of alcohol metabolism may contribute to ADH. NAD, nicotinamide adenine dinucleotide; NADH, reduced nicotinamide adenine dinucleotide; NADP, nicotinamide adenine dinucleotide phosphate. |
This chapter reviews major direct mechanisms of alcohol-induced liver injury, the gut-liver axis, general malnutrition in alcoholic liver disease (ALD), deficiencies of specific nutrients, and nutrition support in ALD. It also highlights how many principles of altered nutrition and nutrition support in ALD apply to other advanced liver diseases (e.g., hepatitis C, nonalcoholic steatohepatitis [NASH]).
DIRECT MECHANISMS OF LIVER INJURY
Ethanol is rapidly removed by the liver as a protective mechanism to prevent CNS depression and injury. However, the toxic metabolic and biochemical processes of ethanol metabolism, including induction of CYP2E1, production of toxic metabolites (e.g., acetaldehyde), and changes of biochemical processes may contribute to the development and progression of ALD.
Induction of CYP2E1
Although the relative contribution of the cytochrome P-450 system to total ethanol metabolism is low, CYP2E1 is strongly induced by alcohol and can contribute to total ethanol metabolism to a far greater extent in alcoholdependent individuals (1). Inhibitors of CYP2E1 partially blocked hepatic injury caused by ethanol in animal models, a finding supporting this hypothesis (2). It is difficult to determine mechanisms in vivo, and, unfortunately, cultured cells exhibit very low activities of CYP enzymes. Therefore, HepG2 cells that overexpress CYP2E1 have been developed. Research using these cells has supported the hypothesis that CYP2E1 is involved in hepatocyte damage from alcohol (3).
The exact mechanisms by which CYP2E1 contributes to ALD still need to be elucidated. However, it is proposed that CYP2E1 contributes to oxidative stress caused by alcohol because this enzyme has been shown to be relatively loosely coupled with cytochrome reductase. CYP2E1 can therefore leak electrons to oxygen to form O2·−, or it can catalyze lipid peroxidation (4). Furthermore, CYP2E1 can bioactivate hepatotoxic agents (e.g., acetaminophen). The induction of this enzyme by chronic ethanol abuse can therefore increase the risk of liver damage by other agents. This enzyme has also been shown to be induced in Kupffer cells. Macrophages overexpressing CYP2E1 respond more robustly to stimulation in culture (5), and this may contribute to the priming effect of alcohol on these cells. Finally, ALD often correlates with an increase in the formation of autoantibodies, and autoantibodies against oxidatively modified CYP2E1 have been detected in the blood of persons with alcoholism (6).
Production of Toxic Metabolites
Acetaldehyde plays a central role in the toxicity of alcohol. Although acetaldehyde is subsequently oxidized to acetate by ALDHs (see Fig. 82.1), the kinetics of this reaction are relatively slow compared with alcohol oxidation, thus allowing acetaldehyde to accumulate detectably in humans consuming alcohol. Several of the systemic toxic effects of ethanol abuse (e.g., flushing, headaches, and nausea) are mediated, at least in part, by direct or indirect effects of elevated acetaldehyde levels. At the more local level, it is proposed that acetaldehyde also plays an etiologic role in ALD (7). For example, acetaldehyde, which is highly unstable, can form adducts with reactive residues on proteins or small molecules (e.g., cysteines), and these chemical modifications can alter or interfere with normal biologic processes and can be directly toxic to the cell.
Modified biologic molecules may also stimulate the host immune system and cause an autoimmune-like response. Antibodies against such oxidatively modified proteins have been found in both humans and animal models of ALD (8, 9). For example, a hybrid adduct of malondialdehyde and acetaldehyde (MAA), unique to alcohol exposure, has been shown to induce an immune response in both humans with alcoholism and animal models of ALD (10). Furthermore, acetaldehyde promotes enhanced glutathione (GSH) utilization and turnover, which results in significant GSH depletion (11). There are at least five different classes of human ADH isoenzymes, based on differences at the molecular level (12). Single nucleotide polymorphisms (SNPs) in the ADH genes that produce functional differences in the kinetic properties of the enzymes and the relative rate of alcohol metabolism have been identified (13). Indeed, correlations between lower ALDH activity and increased risk for ALD have been shown (14). Other products of ethanol metabolism may also be toxic to the liver. In addition to oxidative metabolism,
ethanol can be metabolized via a nonoxidative pathway with fatty acid ethyl esters (FAEEs) as end products (15). These molecules accumulate in the mitochondria and may uncouple oxidative phosphorylation. FAEEs are suspected to contribute to tissue damage in organs that lack oxidative ethanol metabolism (16).
ethanol can be metabolized via a nonoxidative pathway with fatty acid ethyl esters (FAEEs) as end products (15). These molecules accumulate in the mitochondria and may uncouple oxidative phosphorylation. FAEEs are suspected to contribute to tissue damage in organs that lack oxidative ethanol metabolism (16).
Changes in Biochemical Processes
The concentrations of ethanol in the systemic blood can reach quite high levels; for example a BAC of 0.08% equates to an alcohol concentration of approximately 20 mM in the blood. Because of the first-pass effect of the liver on ethanol, the hepatic concentrations of ethanol are much higher than systemic levels. These high alcohol concentrations, coupled with the impressive rate of metabolism by the liver, stress liver cells biochemically. Indeed, although acetate from ethanol oxidation can enter the citric acid cycle after conversion to acetyl-coenzyme A, the various metabolic and biochemical alterations caused by ethanol exposure result in a negative energy balance (11). It is hypothesized that some of these biochemical changes caused by alcohol metabolism may cause ALD.
Steatosis is one of the earliest hepatic changes caused by alcohol, and it was originally thought to be a pathologically inert histologic change. It is now understood that steatosis may play a critical role not only in the initiation but also in the progression of ALD (17, 18). For example, fatty livers are more sensitive to hepatotoxicity from a second hit, such as endotoxin (17). Furthermore, the degree of fatty infiltration is predictive of the severity of later stages of ALD (i.e., fibrosis and cirrhosis) (19, 20). Because the oxidation of ethanol to acetaldehyde by ADH and the subsequent oxidation to acetate by ALDH use NAD+ as an electron acceptor, the ratio of reduced nicotinamide adenine dinucleotide (NADH) to NAD+ is dramatically shifted to a more reduced state. This increase in the reduced state of pyridine nucleotides may also be involved in the accumulation of lipids during alcohol ingestion. Specifically, the shift in the NADH/NAD+ ratio increases the rate of fatty acid synthesis and esterification while simultaneously decreasing mitochondrial β-oxidation of free fatty acids. This change in the redox state can also impair normal carbohydrate metabolism; this has multiple effects, including decreasing the supply of adenosine-5′-triphosphate to the cell (21). Furthermore, investigators have demonstrated that this shift in the pyridine nucleotide redox state can activate the sirtuin family of histone deacetylases (22), which can alter gene expression profiles and thereby indirectly affect the metabolic state of the liver.
Hypoxia
As noted earlier, the oxidation of alcohol by cytochrome P-450 consumes oxygen (see Fig. 82.1). Furthermore, alcohol causes an acute hypermetabolic state in the liver, in which the oxygen consumption rate is doubled (23). This increase in the oxygen extraction rate also increases the intralobular oxygen gradient in the liver (24), thus causing pericentral hypoxia (25, 26). Hypoxia exacerbates the metabolic stress on the liver by further altering the pyridine nucleotide redox state. A decrease in cellular oxygen tension also exacerbates the ethanol-impaired mitochondrial electron flow from decreased delivery of O2 to the mitochondria. After ethanol levels decrease, subsequent reoxygenation can increase prooxidant production via hypoxia or reoxygenation. This effect, in combination with impaired free radical defenses resulting from hypoxia, can contribute to the observed oxidative stress in the liver after alcohol exposure (27, 28).
Oxidative Stress
Reactive oxygen species (ROS) and nitrogen species (RNS) are products of normal cellular metabolism and have beneficial effects (e.g., cytotoxicity against invading bacteria). However, because of the potential of these molecules also to damage normal tissue, the balance between pro-oxidants and antioxidants is critical for the survival and function of aerobic organisms. An imbalance that up-regulates prooxidants or down-regulates antioxidants, thus potentially leading to damage, was termed “oxidative stress” by Sies in 1985 (29). There is evidence that oxidative stress causes ALD (30, 31, 32). Oxidative stress associated with clinical ALD is most likely not solely the result of increased pro-oxidant formation. Persons with alcoholism replace up to 50% of their total daily calories with ethanol (33); therefore, it is not surprising that they have a high rate of nutritional deficiencies. Alcohol abuse also can lead to malabsorption in the gastrointestinal tract, further exacerbating these deficiencies (34). The net effect is that persons with alcoholism often have lower levels of key dietary antioxidant molecules (21), as well as an overall decreased antioxidant status or capacity. Studies in animal models of ALD established a clear link between oxidative stress and the development of experimental liver damage caused by alcohol, and numerous antioxidants were shown to protect against the damaging effects of ethanol in vitro and in vivo models of ALD (35, 36, 37).
The most obvious pathologic changes to the liver during alcohol exposure occur in the hepatocytes. Moreover, the accumulation of products of oxidative stress (e.g., lipid peroxides) is predominantly a hepatocyte event during alcohol administration. This finding indicates that oxidant production by hepatocytes likely plays a key role in alcoholic liver injury. The two major sources of pro-oxidants in hepatocytes are the ethanol-inducible CYP2E1 and mitochondria. The reduction of O2 to H2O by the mitochondrion is not complete and proceeds to O2·− (38). Alcohol exposure even increases the yield of O2·− from hepatocyte mitochondria (39). Elevated pro-oxidant production by mitochondria not only increases the net yield of pro-oxidants in the hepatocyte but also directly damages mitochondrial proteins and DNA. Direct damage can exacerbate mitochondrial aging and stimulate mitochondrial-mediated apoptotic pathways (40). Moreover, alcohol depletes mitochondrial GSH levels (41), a change that increases the response of
hepatocytes to apoptotic stimuli (42). Therefore, it is likely that pro-oxidant production from the mitochondria is critical for the development of severe ALD.
hepatocytes to apoptotic stimuli (42). Therefore, it is likely that pro-oxidant production from the mitochondria is critical for the development of severe ALD.
Inflammation is a key factor in the progression of alcohol-induced liver injury. Both resident (e.g., Kupffer cells) and recruited (e.g., neutrophils and lymphocytes) inflammatory cells are involved in this process. ROS production in hepatocytes is predominantly caused passively by electron leakage from biochemical processes, whereas inflammatory cells are active producers of ROS and RNS. The production of these species is critical for host defense, but these species can also cause damage to normal tissue, if they are inappropriately stimulated.
Inappropriate activation of Kupffer cells plays a key role in the initiation of ALD (43, 44). The production of pro- oxidants is increased in activated Kupffer cells. Major sources of pro-oxidants in these cells include NAD(P) H oxidase and the inducible form of nitric oxide synthase (iNOS or NOS2). Studies have shown that the NAD(P) H oxidase inhibitor, diphenyliodonium (DPI), prevents alcohol-induced liver injury in rats fed alcohol enterally (37). However, pharmacologic inhibitors can have nonspecific effects. Indeed, DPI is likely better defined as a flavoprotein inhibitor than a specific NAD(P)H oxidase inhibitor. However, protection against experimental ALD was also observed in mice deficient in NAD(P)H oxidase [p47phox knockout mice (37)]. Together, these results add weight to the hypothesis that O2·− production from NAD(P)H oxidase plays a key role in the initiation of oxidative stress and experimental ALD. Investigators have demonstrated that iNOS knockout mice (45) and NAD(P)H oxidase-deficient mice (37) are protected against oxidative stress caused by alcohol, a finding indicating that the damaging oxidant in experimental ALD is dependent on the production of O2·− and NO.
Most research into the role of oxidative stress in alcohol-induced liver injury has focused on Kupffer cells or hepatocytes. However, more recent studies indicate that oxidative stress may play a role in the transformation of stellate cells into myofibroblasts, the critical matrix-deposition cell in the fibrotic liver (46). Indeed, oxidant production within the cell may be involved in its transformation (47). It is unclear at this time how much stellate cells contribute to oxidative stress in the organ as a whole, or whether prooxidant production in this cell acts mainly as an autocrine or paracrine signaling event. Because activation of this cell type is key in the progression to fibrosis and cirrhosis in ALD, more research in this area is required.
Oxidative stress can be mediated not only by an increase in ROS and RNS production, but also by a decrease in the antioxidant defenses. For example, alcohol can cause modifications to the cell that may favor oxidative stress. Moreover, persons with alcoholism often have lower antioxidant levels because of nutritional deficiencies. Hypoxia caused by alcohol exposure can impair antioxidant defenses. Additionally, free iron is mobilized by alcohol, and this process can also lead to an increase in transition-metal catalysis to potent oxidants (e.g., the Fenton reaction). Alcohol exposure also inhibits the 26S proteosome in hepatocytes (2) that is responsible for degrading proteins damaged by ROS and RNS. Thus, when this complex is inhibited, proteins damaged by ROS and RNS accumulate, possibly even in the absence of a real increase in pro-oxidants (48). Finally, many proteins and systems are involved in the antioxidant network. This network does not directly block pro-oxidants, but it serves instead as ancillary reductants and maintains the catalytic activity of antioxidant proteins or small molecules. These reactions are energy dependent. Thus, the biochemical stress caused by alcohol exposure can indirectly impair cellular antioxidant defenses.
GUT-LIVER AXIS
Endotoxin or lipopolysaccharide (LPS) is derived from the cell wall of Gram-negative bacteria. Increased endotoxin levels are observed in patients with ALD and in rodent models of ALD. Increased endotoxin levels are also noted in animal models (high-fat or high-fructose feeding) of nonalcoholic fatty liver disease (NAFLD), and endotoxin/gut-derived toxins have been postulated to play an etiologic role in this disease process. Thus, much of what is described related to the gut-liver axis in ALD likely also applies to NAFLD, NASH, and other forms of liver disease. Elevated endotoxin levels in ALD may originate from gram-negative bacterial overgrowth in the intestine, increased intestinal permeability, or impaired hepatic clearance of endotoxin (49). Endotoxin then stimulates the production of tumor necrosis factor (TNF) and other proinflammatory cytokines through Toll-like receptor (TLR-4) signaling, which plays a critical role in the development and progression of ALD (Fig. 82.2). Other bacteria-derived toxins, such as peptidoglycan and flagellin, may also affect TLR signaling and
proinflammatory cytokine production (49). Indeed, injected peptidoglycan increases liver injury or inflammation in alcohol-fed mice compared with control-fed mice, and ethanol feeding increases peptidoglycan levels (49, 50).
proinflammatory cytokine production (49). Indeed, injected peptidoglycan increases liver injury or inflammation in alcohol-fed mice compared with control-fed mice, and ethanol feeding increases peptidoglycan levels (49, 50).
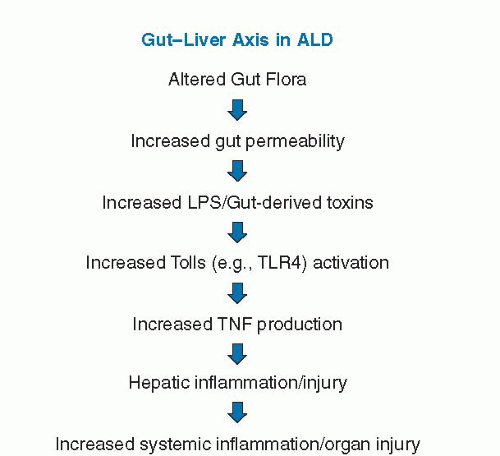 Fig. 82.2. Alterations in gut flora and permeability can lead to activation of hepatic Tolls, subsequent hepatic inflammation or injury, and ultimately systemic inflammation and organ injury. ALD, alcoholic liver disease; LPS, lipopolysaccharide; TLR, Toll-like receptor; TNF, tumor necrosis factor. |
It is generally accepted that the gut flora and gut-derived toxins play a critical role in the development of liver disease and its complications (49, 51, 52, 53, 54, 55, 56, 57). Indeed, in the 1960s, investigators showed that germ-free rodents or rodents treated with antibiotics to “sterilize the gut” were resistant to nutritional and toxin-induced liver injury. Early studies by Broitman et al (53) showed that rats fed a choline-deficient diet developed cirrhosis, which could be prevented by oral neomycin. However, when endotoxin was added to the water supply, neomycin no longer prevented the development of liver injury and fibrosis (53). Subsequently, antibiotics, prebiotics, and probiotics have all been used to prevent experimental alcohol-induced liver injury (58, 59, 60, 61). Increased plasma or hepatic concentrations of proinflammatory cytokines (e.g., TNF-α) were noted in rodent models of ALD, and mice given anti-TNF antibodies or mice lacking TNF-R1 were protected against the development of experimental ALD (62, 63). Moreover, long-term alcohol feeding sensitizes to the hepatotoxicity induced by gut-derived endotoxin and TNF, and specific components of the TLR-4 pathway responsible for alcohol-related liver injury are under active investigation (64, 65). TLR-4 activation by endotoxin results in recruitment of the adaptor molecules MyD88 and Toll/interleukin-1 receptor (TIR) domain-containing adapter inducing interferon-β (TRIF), which each activate separate downstream signaling cascades. Data suggest that the MyD88-independent pathway (TRIF) is more important in the development of experimental ALD, whereas NASH appears to signal through the MyD88-dependent pathway (65). Endotoxin not only plays a role in the fatty liver and liver injury of experimental ALD but also appears to play a role in hepatic fibrosis. Elegant studies from Brenner’s group, using in vitro assays as well as in vivo mixed chimerism studies, showed that endotoxin primes the stellate cell for transforming growth factor-β (TGF-β) stimulated collagen production (66). Thus, LPS also plays a role in fibrosis induction and progression.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


