1Abbreviations: AGE, advanced glycated end product; CCL, chemokine (C-C motif) ligand; CD, Crohn disease; CFU, colony-forming unit; COX, cyclooxygenase; CRP, C-reactive protein; DHA, docosahexaenoic acid; EPA, eicosapentaenoic acid; HLA, human leukocyte antigen; IBD, inflammatory bowel disease; IFN, interferon; Ig, immunoglobulin; IL, interleukin; LOX, lipoxygenase; MCP, monocyte chemoattractant protein; NF-κB, nuclear factor κB; PPAR, peroxisome proliferator-activated receptor; PUFA, polyunsaturated fatty acid; RA, rheumatoid arthritis; RAGE, receptor for advanced glycated end product; STAT, signal transducers and activators of transcription; Th, helper T cell; TNF, tumor necrosis factor; UC, ulcerative colitis.
INFLAMMATION
Inflammation is a normal host defense mechanism that protects the host from infection and other insults. It initiates pathogen killing as well as tissue repair processes and helps to restore homeostasis at infected or damaged sites. Inflammation is typed by redness, swelling, heat, pain, and loss of function. It involves interactions among many cell types and the production of, and responses to, several chemical mediators. Normally, the host is tolerant to microbes and other environmental components that do not pose a threat. This tolerance involves only a limited host response or an active response that is tightly controlled. When an inflammatory response does occur, it is normally well regulated and does not cause excessive damage to the host, is self-limiting, and resolves rapidly. This self-regulation involves the activation of negative feedback mechanisms such as the secretion of anti-inflammatory cytokines, inhibition of proinflammatory signaling cascades, shedding of receptors for inflammatory mediators, and activation of regulatory cells. As such, and controlled properly, regulated inflammatory responses are essential to remaining healthy and maintaining homeostasis.
Pathologic inflammation involves a loss of tolerance or a loss of regulatory processes. When this situation becomes excessive, irreparable damage to host tissues and disease can occur. Typically, diseases or conditions with a well-recognized inflammatory component are treated with general or specific anti-inflammatory pharmaceuticals. However, because many dietary components may influence various elements of inflammation, nutrition may play a role in predisposing to inflammatory conditions, and altered nutrition may be useful in the prevention or therapy of such conditions. This chapter considers the role of inflammation in various diseases and conditions, identifies common mechanisms and markers of inflammation, discusses evidence that selected dietary components can influence inflammatory processes, and indicates the likely mechanisms of action of these dietary components.
General Aspects of the Inflammatory Process
Inflammation may be classified as acute or chronic. Acute inflammation is the initial response of the body to harmful stimuli and is achieved by the increased movement of plasma and leukocytes (especially granulocytes) from the blood into the injured tissues. A cascade of biochemical events propagates and matures the inflammatory response, involving the local vascular system, the immune system, and various cells within the injured tissue. Prolonged inflammation, known as chronic inflammation, leads to a progressive shift in the type of cells present at the site of inflammation and is characterized by simultaneous destruction and healing of the tissue from the inflammatory process. The features of acute and chronic inflammation are compared in Table 63.1. Common to both forms of inflammation are an afferent phase, in which the presence of a “foreign material” is “sensed” by some types of cell, and an efferent phase, in which an inflammatory response is generated to eliminate the perceived hostile intruder. The purpose of the inflammatory response to microorganisms is obvious, and the response is beneficial and necessary to protect the integrity of the body, as long as it does not become unnecessarily destructive or long lasting.
TABLE 63.1 FEATURES OF ACUTE AND CHRONIC INFLAMMATION
ACUTE
CHRONIC
Causative agent
Pathogens, injured tissues
Persistent acute inflammation resulting from nondegradable pathogens, persistent foreign bodies, or autoimmune reactions
Major cells involved
Neutrophils and other granulocytes, mononuclear cells (monocytes, macrophages)
Mononuclear cells (monocytes, macrophages, T lymphocytes, B lymphocytes), fibroblasts
Inflammation caused by nonpathogenic agents can also be beneficial and can remove the foreign material (e.g., by increasing both mucus production and the number of phagocytic cells), but it may also have negative health effects, especially if it has a long duration. Irrespective of the cause of the inflammation, the response involves four major events:
An increased supply of blood to the site of inflammation.
Increased capillary permeability caused by opening of junctions between endothelial cells. This permits plasma and larger molecules, not normally capable of traversing the endothelium, to do so and thus delivers some soluble mediators to the site of inflammation.
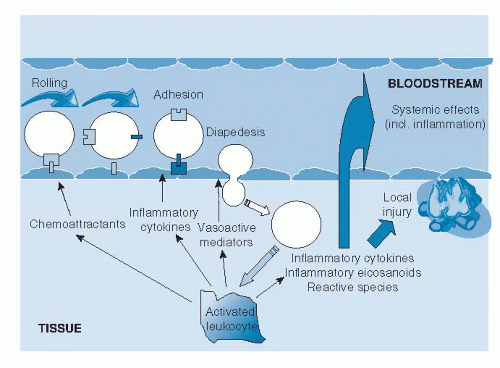
Leukocyte migration from the capillaries into the surrounding tissue (Fig. 63.1) (1). This process is promoted by release of chemoattractants from the site of inflammation and by the up-regulation of adhesion molecules on the endothelium. Once in the tissue, the leukocytes move to the site of inflammation.
4. Release of mediators from leukocytes at the site of inflammation (see Fig. 63.1). These may include lipid mediators (e.g., prostaglandins, leukotrienes), peptide mediators (e.g., cytokines, chemokines), reactive oxygen species (e.g., superoxide), amino acid derivatives (e.g., histamine), and enzymes (e.g., matrix proteases), depending on the cell type, the nature of the inflammatory stimulus, the anatomic site, and the stage during the inflammatory response. These mediators normally would play a role in host defense, but when they are produced inappropriately or in an unregulated fashion, they can damage host tissues and lead to disease. Several of these mediators may act to amplify the inflammatory process by acting, for example, as chemoattractants. Some of the inflammatory mediators may escape the inflammatory site into the circulation and from there exert systemic effects. For example, the cytokine interleukin-6 (IL-6) induces hepatic synthesis of the acute phase protein C-reactive protein (CRP), whereas the cytokine tumor necrosis factor-α (TNF-α) elicits metabolic effects within skeletal muscle, adipose tissue, and bone.
Fig. 63.1. Generalized view of inflammation. (Reproduced with permission from Calder PC, Albers R, Antoine JM et al. Inflammatory disease processes and interactions with nutrition. Br J Nutr 2009;101:S1-45.)
Characteristics of Inflammatory Conditions
Inflammation is a recognized contributor to the pathology of many conditions. In some cases, such as rheumatoid arthritis (RA), inflammatory bowel diseases (IBD), asthma, and psoriasis, the central role of inflammation to the pathologic features is well recognized. Persons with these conditions have heavy infiltration of inflammatory cells at the site of disease activity (e.g., joints, intestinal mucosa, lungs, skin), and they have elevated concentrations of inflammatory mediators at those sites and in the systemic circulation. These conditions are treated with varying levels of success by anti-inflammatory drugs. In other cases, such as atherosclerosis and obesity, the role of inflammation has emerged more recently, and its contribution to the pathologic features alongside the many other relevant factors is less clear. Persons with these conditions show infiltration of inflammatory cells at the site of disease activity (e.g., blood vessel wall, adipose tissue) and have moderately elevated levels of inflammatory mediators in the systemic circulation.
Chronic Inflammation of the Joints: Rheumatoid Arthritis
RA is a common autoimmune disease characterized by chronic inflammation of the synovium of the joints (2). It can lead to long-term joint damage, resulting in chronic pain, loss of function, and disability. The main risk factors for the disease include genetic susceptibility, sex (it is two to three times more common in women than in men), age, smoking, and certain infectious agents. The main predisposing genetic factor is human leukocyte antigen (HLA)-DR4, although other genetic factors have been discovered, such as genetic polymorphisms in the lymphoid protein tyrosine phosphatase (3), which result in altered T-lymphocyte reactivity. In RA, the synovium (or synovial membrane) becomes hypertrophic and edematous. Angioneogenesis, recruitment of inflammatory cells resulting from production of chemokines, local retention, and cell proliferation contribute to the accumulation of cells in the inflamed synovium. Locally expressed degradative enzymes (matrix metalloproteases) digest extracellular matrix and destroy articular structures.
The synovial membrane that extends to the cartilage and bone is known as pannus. It actively invades and destroys the periarticular bone and cartilage at the margin between synovium and bone. T cells are actively involved in the pathogenesis of RA. Activated T cells, which are abundantly present in the inflamed joints of patients with RA, can stimulate other cells (e.g., B cells, macrophages, and fibroblastlike synoviocytes) (4). These T cells are found to participate in the complex network of cell- and mediator-driven events that lead to inflammation and joint destruction. B cells are the source of autoantibodies produced in RA and contribute to immune complex formation and complement activation in the joints (5).
The major effector cells in the pathogenesis of arthritis are synovial macrophages and fibroblasts. Activated macrophages are critical in RA, not only because of macrophage-derived cytokines (in particular, TNF-α and IL-1) in the synovial compartments but also because of their localization at strategic sites within the destructive pannus tissue. Evidence indicates proliferation and expression of inflammatory cytokines and chemokines by fibroblast-like synovial cells in inflamed synovia.
Chronic Inflammation of the Gastrointestinal Mucosa: Inflammatory Bowel Diseases
Ulcerative colitis (UC) and Crohn disease (CD) are the two main forms of IBD. CD can affect any part of the gastrointestinal tract, whereas UC primarily affects the colon (6, 7). IBDs are multifactorial conditions with both genetic and environmental components; the final outcome is driven by an aberrant immune response to normal commensal microbiota in individuals who have a weakened gut epithelial barrier (8).
Although a genetic component is known to be involved in IBD, stronger evidence indicates a genetic link in CD: a mutation in the NOD2/CARD-15 (called IBD-1) gene has been found in 30% of patients with CD (9). NOD2 is a cytoplasmic receptor for certain peptides found in bacterial cell walls that may reduce the ability of patients with CD who have this mutation to clear invasive bacteria. Indeed, evidence indicates microbial involvement in both forms of IBD, with disturbed interaction between the mucosal immune system and the commensal gut microbiota. In both forms of IBD, large infiltrates of neutrophils are present in the inflamed tissue. The T-cell response profiles associated with UC and CD are different in that a helper T-cell (Th1) pattern of cytokine formation develops in CD with increased production of TNF-α, interferon (IFN)-γ, IL-12, IL-6, and IL-1β, whereas UC more resembles a modified Th2 profile in which cytokines including IL-5 and IL-10 are up-regulated, although IL-4 is not. In addition to this change in cytokine profile, intestinal B lymphocytes produce large amounts of immunoglobulin G (IgG). TNF-α is expressed in the intestinal mucosa of patients with IBD and triggers inflammation through a nuclear factor κB (NF-κB)-dependent signaling cascade.
Many of the cytokines involved act on the signal transducers and activators of transcription (STAT) family. STAT-3 signaling has been found in UC and CD, in which it has been shown to be confined to areas of active inflammation, infiltrating macrophages, and T cells. STAT-3 induces transcription of the proinflammatory cytokine IL-6, which can increase resistance of T cells to apoptosis and lengthen the chronicity of CD as a result of the accumulation of reactive T cells. Other factors implicated in CD include generation of matrix metalloproteinases, which can degrade extracellular matrices and cause ulceration and tissue destruction.
Chronic Inflammation of the Airways: Asthma
Asthma, a chronic inflammatory disease of the lungs, is traditionally classified as allergic or nonallergic. Allergic asthma is the most common form in children, whereas in adults, asthma without known allergen triggers is more common. However, the distinction depends on the demonstration of triggering allergens and is somewhat unclear. Various “nonspecific” irritants may aggravate asthma and trigger an asthmatic attack.
Asthma has chest tightness, wheeze, cough, and dyspnea as prominent symptoms; and it is functionally characterized as reversible bronchial obstruction, caused by contraction of the smooth muscle layer in the mucosa of the bronchi, by mucus production, mucosal edema, and mucosal inflammation. Airway hyperresponsiveness (oversensitivity and overreactivity to stimuli) is typically present in asthma.
A prominent cell in the asthmatic inflammation is the eosinophil, together with lymphocytes. Granulocytes other than eosinophils may be present to varying degrees. The inflammation may lead to destruction and shedding of the epithelial cell layer. Over time, structural changes take place in asthma—so-called remodeling. Inflammation becomes permanent and more severe, and reversibility of the airway obstruction is less complete.
Several genes have been implicated in asthma (e.g., ADAM33) (10). Investigators have estimated that more than a dozen polymorphic genes regulate features of asthma such as the inflammatory response, IgE synthesis, cytokine and chemokine production, airway remodeling, and airway function (11). At the heart of the allergic reaction is the interaction between IgE molecules bound to specific receptors on the membrane of mast cells and their corresponding allergens. When the IgE molecules are cross-linked by allergen, the mast cell is triggered to release the potent inflammatory mediators contained in its cytoplasmic granules, and the allergic inflammatory response develops. This response has two phases: an early virtually immediate reaction and a late response developing after several hours. Mast cells are the key cells in the early response, whereas eosinophils are the predominant cell in the late response. Increased levels of the Th2 cytokines IL-4, IL-5, IL-9, and IL-13 have been demonstrated in the asthmatic airway (12). This Th2-driven inflammation has two arms: one through B cells activated by IL-4 to produce IgE, which triggers the mast cell-mediated allergic inflammation; and the other through IL-4, but mainly by IL-13-mediated direct effects on epithelium and bronchial smooth muscle (13). TNF-α has also been reported to play an important role in severe asthma (14).
Chronic Inflammation of the Skin: Psoriasis
Psoriasis is a common inflammatory disease of the skin, although joint symptoms can also be a feature. A genetic susceptibility and associations with other inflammatory conditions are known. Streptococcal infections and physical trauma to the skin may also be involved. The pathophysiology involves an interaction between the immune system and the skin. Psoriasis is characterized by an infiltrate of T lymphocytes into the dermis, formation of clusters of neutrophils in the epidermis, involvement of two or three layers of the epidermis in proliferation, and greatly accelerated but incomplete differentiation. Activation of the innate immune system by streptococcal products and, most likely, as yet unidentified factors induces release of cytokines including IFN-α and IFN-γ. The cellular source of these cytokines is unclear, but it may be dendritic cells. These cytokines activate keratinocytes to proliferate and to produce angiogenic factors that induce proliferation of dermal microvessels.
Chronic Inflammation of the Vascular Wall: Atherosclerosis
Atherosclerosis or “hardening of the arteries” is the major cause of cardiovascular disease. Endothelial dysfunction is the key underlying event, and this is characterized by altered endothelial function, enhanced adhesion molecule expression, and impaired endothelium-dependent vasodilator responses. Leukocytes become attached to the dysfunctional endothelium and subsequently accumulate within the subendothelial space. Monocyte-derived macrophages are converted to lipid-laden foam cells within the artery wall, thus giving rise to a lesion termed the fatty-streak. The conversion of the fatty streak into a fibrous atherosclerotic plaque necessitates the recruitment and proliferation of vascular smooth muscle cells (15).
Atherosclerosis is now considered to be a chronic inflammatory disease, and at every stage of its evolution it is characterized by monocyte-macrophage and T-lymphocyte infiltration (16). The possible stimuli to this inflammatory process include oxidized low-density lipoproteins, homocysteine, free radicals generated from cigarette smoking, and infectious microorganisms. The T-cell infiltrates are predominantly T-helper (i.e., CD4+) cells, and cells derived from human lesions react to antigens derived from oxidized low-density lipoproteins, heat shock proteins, and microorganisms (16). The cytokine milieu within atherosclerotic lesions is thought to promote a Th1-dominated response associated with macrophage activation and the production of IFN-γ and IL-1β. The ongoing inflammation involves various growth factors and cytokines, which lead to intimal thickening by stimulating smooth muscle cell migration, proliferation, and extracellular matrix generation.
Chronic Inflammation of Adipose Tissue: Obesity
Obesity is characterized by an expansion of the mass of adipose tissue and dramatic changes in its distribution in the body. A mechanistic link between obesity and lowgrade inflammation was first proposed by Hotamisligil et al (17), who showed that white adipose tissue synthesizes and releases TNF-α. The range of inflammatory proteins produced by adipose tissue is now known to be extremely wide and includes leptin, adiponectin, some acute phase proteins, cytokines (including IL-1, IL-6, and TNF-α), chemokines (including IL-8, monocyte chemoattractant protein-1 [MCP-1], RANTES [now known as chemokine (C-C motif) ligand CCL5], and macrophage inflammatory protein-1α and -1β [now known as CCL3 and CCL4, respectively]), and complement factors (including C3) (18). Obesity is associated with chronic elevation of the circulating concentrations of inflammatory proteins including several acute phase inflammatory proteins such as CRP, proinflammatory and anti-inflammatory cytokines, and soluble adhesion molecules (18).
Adipose tissue is a heterogeneous tissue composed of several cell types: mature adipocytes, preadipocytes, fibroblasts, endothelial cells, mast cells, granulocytes, lymphocytes, and macrophages. Because of the heterogeneity of cells in the adipose tissue, the cellular source of the inflammatory factors secreted by the tissue into the circulation remains unknown. However, both adipocytes and classic inflammatory cells, especially macrophages, seem likely to be involved. T lymphocytes appear to play a key early role in adipose tissue inflammation (19). Many mediators synthesized by the adipose tissue are candidates to attract inflammatory cells. Leptin induces adhesion proteins, thus facilitating the migration of monocytes. Conversely, adiponectin may inhibit this process. MCP-1 is a strong chemoattractant and is thought to be a major player in macrophage accumulation within the adipose tissue. Local hypoxia could also play an important role in the attraction and retention of macrophages within the adipose tissue.
Common Features of Chronic Inflammatory Processes
Although inflammation-induced tissue damage occurs in an organ-specific manner (joints, gut, lungs, skin, blood vessel wall, adipose tissue) in different diseases or conditions, some commonality exists among the responses seen in the different organs (summarized in Table 63.2). In general, the inflammatory response observed is normal, but it occurs in the wrong context; this relates to inappropriate barrier function (epithelial or endothelial), inappropriate triggering (i.e., a response to a normally benign stimulus equivalent to a loss of tolerance), lack of down-regulation to control the response, and tissue destruction with a loss of function. In some cases, the inflammation is the result of exogenous triggers such as allergens or microbes. In other cases, it is secondary to tissue damage caused by endogenous molecules such as oxidized low-density lipoprotein.
TABLE 63.2 SUMMARY OF THE CHARACTERISTICS OF INFLAMMATORY DISEASE STATES
Modified with permission from Calder PC, Albers R, Antoine JM et al. Inflammatory disease processes and interactions with nutrition. Br J Nutr 2009;101:S1-45.
The involvement of different triggers is also reflected in the distinct associations with polymorphisms in receptors involved in pattern recognition such as NOD2 in CD or with other molecules involved in specific adaptive immune responses such as HLA-DR subtypes in UC and RA (see Table 63.2). However, although trigger, localization, and resulting clinical symptoms are different, many of the processes, cells, and molecules involved in the actual inflammatory response are remarkably similar (see Table 63.2).
Most, if not all, of the chronic inflammatory diseases considered here are characterized by overproduction of cytokines (TNF-α, IL-1β, IL-6, IFN-γ), chemokines (IL-8, MCP-1), eicosanoids (prostaglandin E2, 4-series leukotrienes), and matrix metalloproteinases. Elevated levels of these mediators act to amplify the inflammatory process (e.g., by attracting further inflammatory cells to the site) and contribute to tissue destruction (see Fig. 63.1) and to the clinical symptoms observed. Many of these mediators are positively regulated through NF-κB, and some are negatively regulated through peroxisome proliferator-activated receptors (PPARs) and liver X receptors. Entry of inflammatory cells to sites of inflammatory activity is facilitated by up-regulation of adhesion molecules on the endothelium, a process that is promoted by inflammatory cytokines and by a range of inflammatory triggers, frequently acting through NF-κB. The continuous process of tissue injury, healing, and repair, in response to the release of cytokines, chemokines, and growth factors by infiltrating inflammatory cells, as well as resident tissue cells, results in tissue remodeling.
Only gold members can continue reading. Log In or Register to continue