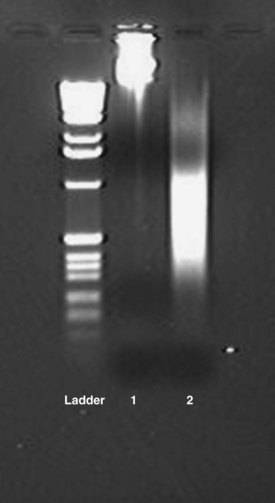
Chapter 39 Rossa W.K. Chiu, M.B.B.S., Ph.D., F.H.K.A.M.(Pathology), F.R.C.P.A. and Y.M. Dennis Lo, M.A., D.M., D.Phil., F.R.C.P.,(Lond. & Edin.), F.R.C.Path., F.R.S.* Many of the key advances in our understanding of DNA and RNA have been achieved by the study of purified nucleic acids. In a molecular diagnostic laboratory, some tests can be performed directly on biological fluids, but techniques for isolation of nucleic acids often are critical. Nucleic acid isolation refers to the process of separating DNA or RNA from its surrounding materials; nucleic acid extraction is the technique of removing DNA or RNA from surrounding materials. With the advent of the polymerase chain reaction (PCR), molecular analyses could be performed on a variety of specimens, including whole blood, plasma, serum, tissue biopsies, cultured cells, buccal swabs, cerebrospinal fluids, amniotic fluids, and paraffin-embedded tissues. Isolation of DNA and/or RNA is usually a key preliminary step in molecular analysis.43 DNA- and RNA-specific nucleases (DNases and RNases, respectively) can degrade DNA and RNA molecules in biological specimens. Hence, specimens should be properly preserved before molecular analyses. DNA molecules are generally stable in specimens when stored at −80 °C. However, RNA molecules are much more fragile than DNA. This is because, unlike DNA, RNA is not protected by a stable double-helical conformation, and RNA is subject to alkaline hydrolysis via the 2′ hydroxyl group of its ribose moiety. In addition, RNases are ubiquitous and difficult to inactivate or denature. Hence, special attention should be paid when one undertakes operations on RNA.12 Tissue specimens intended for RNA analysis should be processed promptly upon collection. One preservation method is to snap-freeze the specimens by liquid nitrogen. However, snap-frozen tissues are laborious to handle in the subsequent homogenization step (see later). For example, to minimize the exposure of RNA to RNases, the frozen tissues should be ground to a fine powder before denaturants are added. Moreover, to prevent the release of RNases from thawing cells, the tissues should be kept frozen during the grinding process; thus liquid nitrogen has to be added at intervals. Alternatively, RNA can be protected by incubating tissue specimens in preservation agents such as RNAlater (Ambion/Applied Biosystems, Austin, Tex) and PrepProtect Stabilization Buffer (Miltenyi Biotec Inc., Auburn, Calif).33 RNA molecules in such preserved tissues are stable at room temperature for up to a week.22 Specimen handling and transportation procedures are thereby facilitated. The preserved tissues can be directly homogenized (see later) without further processing. Blood is the most common specimen type in a diagnostic laboratory because it is more readily available than a biopsy specimen. During peripheral blood collection and processing, ex vivo changes in gene expression and RNA degradation continue to occur, and can affect subsequent analyses.38 To overcome such problems, vacutainer tubes prefilled with preservation agents, such as those in the PAXgene Blood RNA System (PreAnalytiX GmbH, Hombrechtikon, Switzerland), are available for immediate stabilization of RNA profiles during blood collection.50 Plasma and serum are routinely used for circulating nucleic acid studies (see Chapter 45). Plasma DNA has been shown to be stable in unprocessed whole blood for up to 24 hours at room temperature.45 Plasma RNA has also been shown to be stable in unprocessed whole blood when collected in tubes containing ethylenediaminetetraacetic acid (EDTA) for up to 24 hours at 4 °C.45 However, in contrast to short-term storage, the concentration of circulating DNA in harvested serum specimens has been reported to decrease by 0.66 copies per milliliter for each month of storage.25 More marked degradation upon long-term storage was observed for plasma RNA.49 To protect RNA in plasma and serum during storage, several investigators have suggested mixing RNase-inhibitory agents, such as TRIzol LS (Invitrogen, Life Technologies, Carlsbad, Calif), with specimens before storage at −80 °C.45,49 In urine specimens, bacterial and fungal contamination as well as calcium oxalate crystal formation can influence the yield of downstream nucleic acid isolation. Hence, long-term storage of urine specimens with EDTA at −80 °C has been recommended to resolve contamination problems and to inhibit any possible nuclease activity in urine.20,31,42 Homogenization refers to the breakdown of tissue into small fragments by mechanical or enzymatic means. Complete homogenization is essential for isolating nucleic acids with high quality and yield. For DNA isolation from soft tissues, homogenization is usually performed by incubating the specimens with digestion enzymes such as protease K at 56 °C overnight.51 On the other hand, the roto/stator method is preferred for RNA isolation. This method involves mechanical disruption of tissues in lysis buffer, composed of phenol, sodium dodecyl sulfate, or guanidine salts (e.g., TRIzol from Invitrogen). The buffers not only lyse cells directly, they also inhibit RNase effectively during homogenization.9 Solid-phase extraction methods are more robust and generally are based on the principle of DNA adsorption onto silica in the presence of chaotropic salts, such as guanidine thiocyanate, and ethanol.3,37 The silica is typically coated onto membrane filters or magnetic particles. Silica-impregnated filters that are housed in plastic columns are the most commonly adopted format. DNA binds reversibly with silica, depending on the ionic strength of the environment.41 After cell lysis and protein denaturation, DNA is precipitated by the addition of ethanol. The solution is then passed through the silica-impregnated filter, which binds and purifies DNA from other debris present in the ethanol solution. Centrifugation or vacuum manifolds can be used for the filtration step. The bound DNA is washed and subsequently eluted using nuclease-free water or low ionic strength buffers. Similarly, methods based on the use of silica-coated magnetic particles allow the isolation of ethanol-precipitated DNA by placing the solution under a magnetic field whereby DNA molecules adsorbed to the magnetic beads are retained. The DNA is subsequently eluted from the beads in low ionic strength buffer or water. The underlying principles of RNA extraction are essentially identical to those of DNA isolation. Both solution- and solid-phase extraction protocols are available. The most commonly used solution-based method is guanidine thiocyanate-phenol-chloroform extraction.9,10 Guanidine isothiocynate is a chaotropic agent used for protein denaturation while RNase activity is inhibited. In a mixture of guanidine isothiocynate, phenol (e.g., TRIzol from Invitrogen), sodium acetate, and chloroform in an acidic condition, total RNA is partitioned into the aqueous phase, and DNA molecules and proteins remain in the interphase and the organic phase, respectively. Total RNA in the aqueous phase is next precipitated by the addition of isopropanol. For solid-phase RNA extraction protocols, lysis/binding buffer includes chaotropic salts, such as guanidine isothiocyanate, to protect RNA. The ionic strength of the buffer is optimized to promote the binding of RNA molecules onto the silica matrix. For specimens with high protein and lipid contents, such as plasma and serum, agglutination of lysed samples with the silica matrix can occur. In addition, genomic DNA present in the specimens competes with the RNA for binding to the silica matrix. These factors can adversely affect the isolation of RNA species that exist at trace amounts in the specimens. One such example is the extraction of cell-free fetal RNA in maternal plasma. To overcome these limitations, several investigators have developed a combined solution- and solid-phase RNA extraction protocol.34,49 As an initial step, RNA molecules are separated from contaminants such as protein, lipid, and DNA by the TRIzol protocol. RNA molecules contained in the aqueous phase are then isolated by the silica-column method. The guanidine thiocyanate-phenol-chloroform protocols are relevant to the isolation of total RNA, which comprises messenger RNA (mRNA), ribosomal RNA (rRNA), transfer RNA (tRNA), and small nuclear RNA. Protocols have been developed for the specific isolation of mRNA; these are based on capture of polyadenylated tails on the 3′ ends of mRNA by hybridization to cellulose-bound oligo(dT) molecules.12 It is worth noting that most of the commercial silica-based kits are inefficient in extracting small nucleic acid species, such as microRNA. Because the binding efficiency of nucleic acids to the silica matrix is dependent on salt concentrations, pH, and the ethanol content of the washing buffer in a size-dependent manner, most commercial kits have optimized these parameters to isolate nucleic acids larger than 150 nucleotides. Hence, microRNA extraction should be performed by using the solution-based method (e.g., TRIzol protocol) or with a silica-column protocol that has been optimized for binding microRNA molecules (e.g., mirVana from Ambion).32 Nucleic acid molecules absorb ultraviolet light maximally at a wavelength of 260 nm owing almost entirely to the constituent bases. Thus, DNA or RNA yield can be quantified by spectrophotometric measurement of absorbance at 260 nm, with higher absorbance values indicating higher yield. For example, a solution containing 50 mg/L of pure double-stranded DNA has an absorbance of 1.0 at 260 nm. Purity can be evaluated by assessing the ratio of spectrophotometric absorbances at 260 nm and 280 nm (A260/A280). Absorbance ratios greater than 1.8 indicate minimal contamination with proteins. The sizes of the isolated genomic DNA can be estimated by gel electrophoresis. Good quality DNA extractions are generally associated with higher molecular weight fragments (Figure 39-1). Similarly, total RNA integrity can be assessed by estimating the size distribution of the extracted RNA and the appearances of the 18S and 28S rRNA peaks (Figure 39-2). Total RNA preparations of high quality generally have a 28S/18S peak ratio greater than 2.0. With RNase degradation, the RNA size distribution is shifted toward the smaller fragments with a reduction in the 28S/18S rRNA ratio (see Figure 39-2).
Nucleic Acid Isolation
Specimen Preservation
Tissue Homogenization and Cell Lysis
DNA Isolation
RNA Isolation
Assessment of Nucleic Acid Yield and Quality
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Basicmedical Key
Fastest Basicmedical Insight Engine