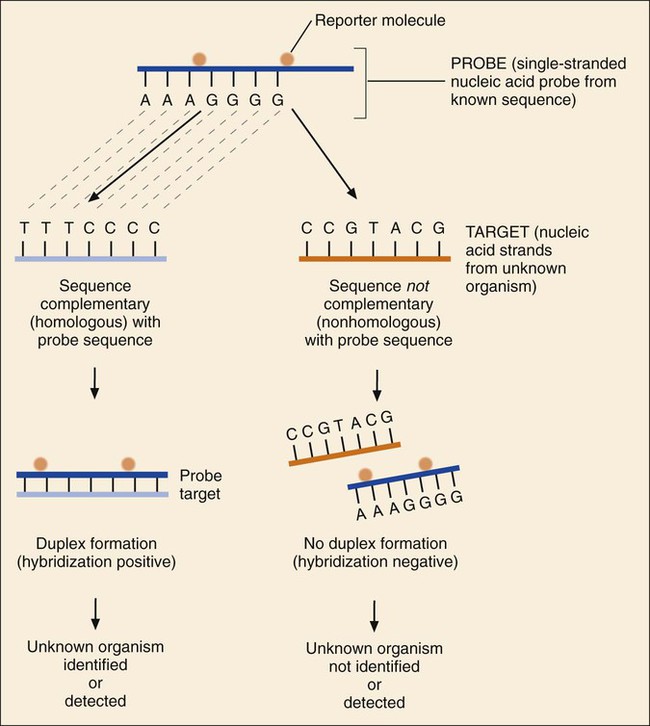
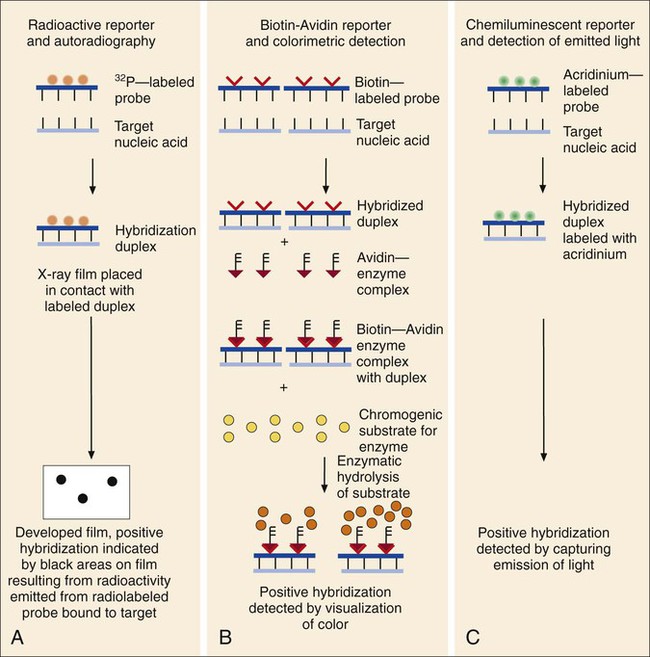
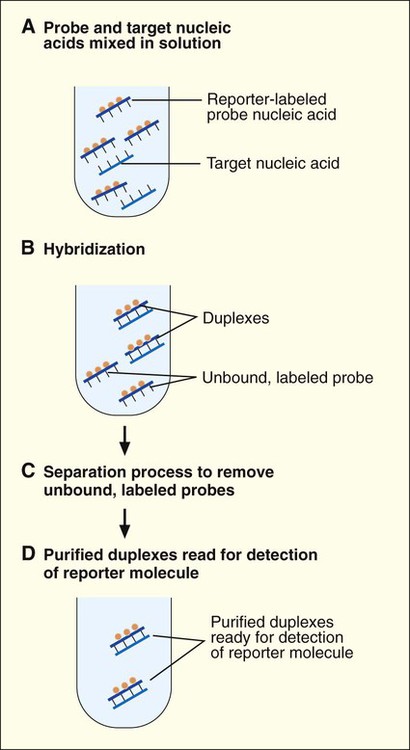
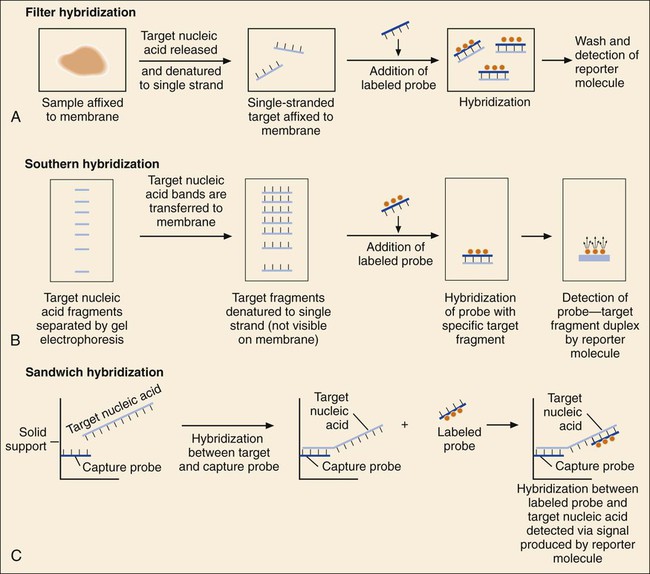
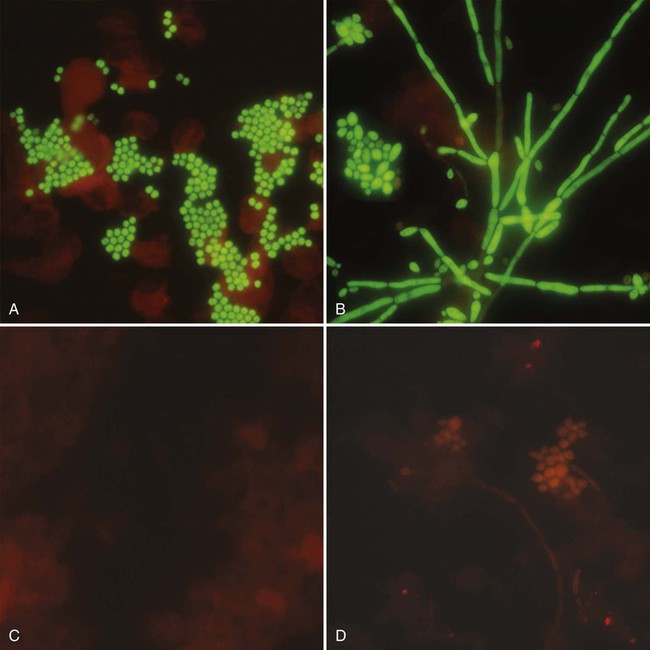
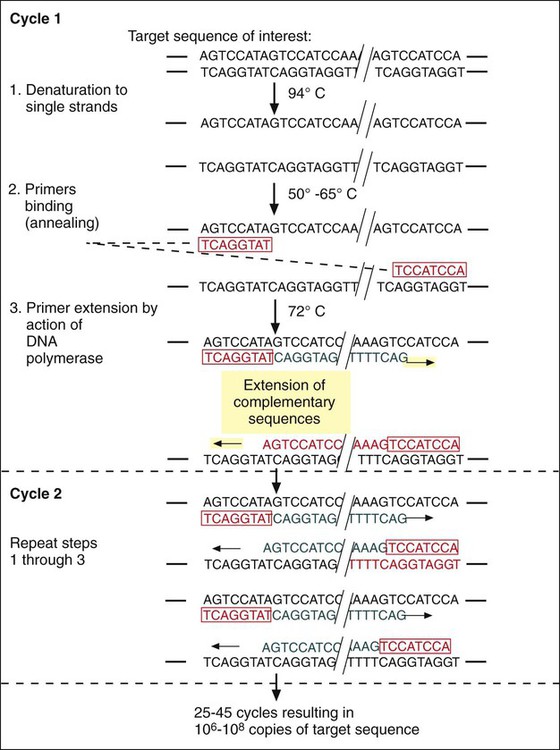
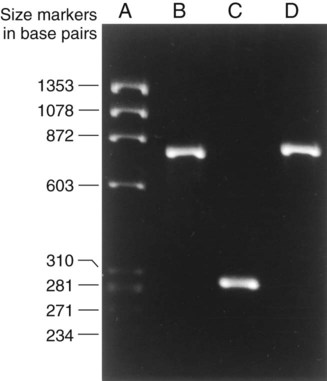
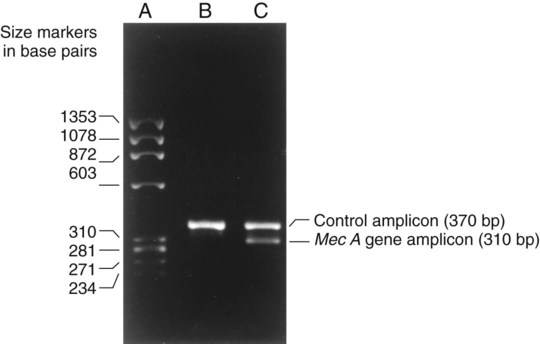
1. Explain the importance of molecular testing in the microbiology laboratory. Also, list the three categories of molecular testing and provide a brief explanation of the methodology for each type. 2. Outline the four-step process in nucleic acid hybridization. 3. Explain the methodology for peptide nucleic acid fluorescent in situ hybridization (PNA FISH) and provide an example of a clinical application. 4. List the three types of nucleic acid extraction; also, compare and contrast the advantages, disadvantages, and outcomes for each. 5. Compare direct molecular hybridization detection with amplified direct detection. 6. Outline the three major steps in polymerase chain reaction (PCR) and describe the critical parameters of each step, including reagents, temperature, time, and interfering substances. 7. Define reverse transcription polymerase chain reaction (RT-PCR); also, explain how and why it is used and the methodology that differentiates it from a traditional PCR test. 8. Explain real-time PCR and list the four potential advantages this procedure has over conventional PCR. 9. Define palindrome, blunt and staggered cuts, and restriction endonuclease. 10. Describe how restriction endonucleases are used in epidemiologic applications and strain typing in molecular diagnostics. 11. Define pulsed-field gel electrophoresis (PFGE) and restriction fragment length polymorphism (RFLP) and state an application for each. The principles of bacterial cultivation and identification discussed in Chapter 7 focus on phenotypic methods. These methods analyze readily observable bacterial traits and “behavior.” Although these strategies are the mainstay of diagnostic bacteriology, notable limitations are associated with the use of phenotypic methods. These limitations are as follows: • Inability to grow certain fastidious pathogens • Inability to maintain viability of certain pathogens in specimens during transport to laboratory • Extensive delay in cultivation and identification of slowly growing pathogens • Lack of reliable methods to identify certain organisms grown in vitro • Use of considerable time and resources in establishing the presence and identity of pathogens in specimens The explosion in molecular biology over the past 20 years has provided alternatives to phenotypic strategies used to identify organisms in the diagnostic microbiology laboratory. These alternatives have the potential to avert some of the aforementioned limitations. Applications of molecular diagnostics in microbiology provide for the qualitative and quantitative detection of organisms, microbial identity testing, and genotyping for drug resistance. The detection and manipulation of nucleic acids (deoxyribonucleic acid [DNA] and ribonucleic acid [RNA]) allows microbial genes to be examined directly (i.e., genotypic methods) rather than by analysis of their products, such as enzymes (i.e., phenotypic methods). Additionally, non–nucleic acid–based analytic methods that detect phenotypic traits undetectable by conventional strategies (e.g., cell wall components) have been developed to enhance bacterial detection, identification, and characterization. For laboratory diagnosis of infectious diseases to remain timely and effective, strategies that integrate conventional, nucleic acid–based, and analytic techniques must continue to evolve. Because molecular diagnostic tests are based on the consistent and somewhat predictable nature of DNA and RNA, understanding these methods requires a basic understanding of nucleic acid composition and structure. Therefore, a review of the section Nucleic Acid Structure and Organization, in Chapter 2, is recommended. Hybridization methods are based on the ability of two nucleic acid strands with complementary base sequences (i.e., they are homologous) to bond specifically with each other and form a double-stranded molecule, also called a duplex or hybrid. This duplex formation is driven by the hydrophobic structure and hydrogen bonding pattern of the nucleotides, which ensure that the base adenine always bonds to thymine (two hydrogen bonds), whereas the bases guanine and cytosine (three hydrogen bonds) always form a bonding pair (see Figure 2-2). Because hybridization requires nucleic acid sequence homology, a positive hybridization reaction between two nucleic acid strands, each from a different source (i.e., intermolecular), indicates genetic relatedness between the two organisms that donated each of the nucleic acid strands for the hybridization reaction. Hybridization reactions may also occur within the same molecule (intramolecular hybridization). Intramolecular hybridization is used to differentiate sequences with electrophoretic separation. Hybridization assays require detection or identification of two nucleic acid strands; one strand (the probe) originates from an organism or nucleic acid sequence of known identity, and the other strand (the target) originates from an unknown organism (Figure 8-1). Positive hybridization identifies the unknown organism as being the same as the probe-source organism or sequence. With a negative hybridization test result, the organism remains undetected or unidentified. The single-stranded nucleic acid components used in hybridization may be either RNA or DNA; therefore, DNA-DNA, DNA-RNA, and even RNA-RNA duplexes may form, depending on the specific design of the hybridization assay. Hybridization assays may be classified as either nonamplified or amplified. A nonamplified assay requires three steps: preparation of the test sample (nucleic acid), hybridization, and signal detection. Amplified assays include an additional step; initial hybridization is followed by a target amplification and then by signal detection. Amplified assays allow detection of as little as a single organism or nucleic acid sequence in the sample material. The basic steps in a hybridization assay include: 1. Production and labeling of single-strand nucleic acid probe 2. Preparation of single-strand target nucleic acid 3. Mixture and hybridization of target and probe nucleic acid All hybridization tests must have a means to detect or measure the hybridization reaction. This is accomplished with the use of a “reporter” molecule attached to the single-stranded nucleic acid probe. Probes may be labeled with a variety of molecules, but most commonly, radioactive (e.g., 32P, 3H, 125I, or 35S), biotin-avidin, digoxigenin, fluorescent, or chemiluminescent labels are used (Figure 8-2). Radioactive labels are directly incorporated through chemical modification into the probe molecule. With the use of radioactively labeled probes, hybridization is detected by the emission of radioactivity from the probe-target complex (see Figure 8-2, A). Quantification of the complexes may be achieved through scintillation counting or densitometry. Although this is a highly sensitive method for detecting hybridization, the requirements for radioactive training, monitoring, licensing, and disposal of radioactive waste have limited the use of radioactive labeling in the diagnostic setting. Biotinylation is a nonradioactive alternative for labeling nucleic acid probes that involves the chemical incorporation of biotin. Biotin labels are classified as indirect, based on the need for a secondary complex formation. Biotin-labeled probe-target nucleic acid duplexes are detected using avidin, a biotin-binding protein conjugated with an enzyme, such as horseradish peroxidase. When a chromogenic substrate is added, the peroxidase produces a colored product that can be detected visually or spectrophotometrically (see Figure 8-2, B). Chemiluminescent reporter molecules can be chemically linked directly to the nucleic acid probe without using a conjugated antibody. These molecules (e.g., acridinium or isoluminol) emit light during hybridization between the chemiluminescent-labeled probe and target nucleic acid. The light is detected using a luminometer (see Figure 8-2, C). • Salt concentration in the hybridization buffer (stringency increases as salt concentration decreases) • Temperature (stringency increases as temperature increases) • Concentration of destabilizing agents (stringency increases with increasing concentrations of formamide or urea) The method of detecting hybridization depends on the reporter molecule used for labeling the probe nucleic acid and on the hybridization format (see Figure 8-2). Hybridization using radioactively labeled probes is visualized after the reaction mixture is exposed to radiographic film (i.e., autoradiography). Hybridization with nonradioactively labeled probes is detected using colorimetry, fluorescence, or chemiluminescence, and detection can be somewhat automated using spectrophotometers, fluorometers, or luminometers, respectively. The more commonly used nonradioactive detection systems (e.g., digoxigenin, chemiluminescence, fluorescence) are able to detect approximately 104 target nucleic acid sequences per hybridization reaction. Hybridization reactions can be done using either a solution format or solid support format. In the solution format, probe and target nucleotide strands are placed in a liquid reaction mixture that facilitates duplex formation; hybridization occurs substantially faster than with a solid support format. However, before duplex formation can be detected, the hybridized, labeled probes must be separated from the nonhybridized, labeled probes (i.e., “background noise”). Separation methods include enzymatic digestion (e.g., S1 nuclease) of single-stranded probes and precipitation of hybridized duplexes, use of hydroxyapatite or charged magnetic microparticles that preferentially bind duplexes, or chemical destruction of the reporter molecule (e.g., acridinium dye) attached to unhybridized probe nucleic acid. After the duplexes have been “purified” from the reaction mixture and the background noise minimized, hybridization detection can proceed by the method appropriate for the type of reporter molecule used to label the probe (Figure 8-3). Filter (membrane) hybridization has several variations. Filter hybridizations are often referred to as “dot blots.” The target sample, which can be previously purified DNA, the microorganism containing the target DNA, or the clinical specimen containing the microorganism of interest, is affixed to a membrane (e.g., nitrocellulose or nylon fiber filters). To identify specimens, samples are usually oriented on the membrane using a template or grid. The membrane is chemically treated, causing release of the target DNA from the microorganism and denaturing the nucleic acid to single strands. The membrane is then submerged in a solution containing labeled nucleic acid probe and incubated, allowing hybridization to occur. After a series of incubations and washings to remove unbound probe, the membrane is processed for detection of duplexes (Figure 8-4, A). An advantage of this method is that a single membrane can hold several samples for exposure to the same probe. Southern hybridization is another method that uses membranes as the solid support. In this instance, the nucleic acid target is purified from the organisms and digested with specific enzymes to produce several fragments of various sizes (Figure 8-4, B) (also see Enzymatic Digestion and Electrophoresis of Nucleic Acids later in this chapter). The nucleic acid fragments, which carry a net negative charge, are subjected to an electrical field, forcing them to migrate through an agarose gel matrix (i.e., gel electrophoresis). Because fragments of different sizes migrate through the porous agarose at different rates, they can be separated by molecular size. When electrophoresis is complete, the nucleic acid fragments are stained with the fluorescent dye ethidium bromide so that fragment “banding patterns” can be visualized on exposure of the gel to ultraviolet (UV) light. For southern hybridization, the target nucleic acid bands are transferred to a membrane that is submerged in solution, allowing for hybridization of the nucleic acid probe. After hybridization, the southern hybridization membrane is used to detect the specific target nucleic acid fragment carrying the base sequence by using radiolabeled, fluorescent, or substrate-labeled detection. The complexity, time, and labor intensity of the procedure precludes its common use in most diagnostic settings. With sandwich hybridizations two probes are used. One probe is attached to the solid support, is not labeled, and via hybridization “captures” the target nucleic acid from the sample to be tested. The presence of this duplex is then detected using a labeled second probe that is specific for another portion of the target sequence (Figure 8-4, C). Sandwiching the target between two probes decreases nonspecific reactions but requires a greater number of processing and washing steps. For such formats, plastic microtiter wells coated with probes have replaced filters as the solid support material, thereby facilitating the use of these multiple-step procedures for testing a relatively large number of specimens. PNA probes are synthetic pieces of DNA that have unique chemical characteristics in which the negatively charged sugar-phosphate backbone of DNA is replaced by a neutral polyamide backbone of repetitive units (Figure 8-5). Individual nucleotide bases can be attached to this neutral backbone, which then allows the PNA probe to hybridize to complementary nucleic acid targets. Because of the synthetic structure of the backbone, these probes have improved hybridization characteristics, providing faster and more specific results than traditional DNA probes. In addition, because these probes are not degraded by ubiquitous enzymes, such as nucleases and proteases, they provide a longer shelf-life in diagnostic applications. PNA FISH is a novel fluorescent in situ hybridization (FISH) technique that uses PNA probes to target species-specific ribosomal RNA (rRNA) sequences. Upon penetration of the microbial cell wall, the fluorescent-labeled PNA probes hybridize to multicopy rRNA sequences within the microorganisms, resulting in fluorescent cells. Recently, AdvanDx (Woburn, Massachusetts) introduced in vitro diagnostic kits (using PNA FISH), which have been approved by the U.S. Food and Drug Administration (FDA). These kits can be used to directly identify S. aureus and C. albicans and to differentiate Enterococcus faecalis from other enterococci in blood cultures. In brief, a drop from a positive blood culture bottle is added to a slide containing a drop of fixative solution. After fixation, the fluorescent-labeled PNA probe is added and allowed to hybridize; slides are washed and air dried. After the addition of a mounting medium and a coverslip, the slides are examined under a fluorescent microscope using a special filter set. Identification is based on the presence of bright green, fluorescent-staining organisms (Figure 8-6, A and B). For negative results, only slightly red-stained background material is observed (Figure 8-6, C and D). Multiple studies have been done to evaluate the efficacy of the PNA FISH kits for identifying S. aureus and C. albicans in positive blood cultures. The kits have demonstrated high sensitivity and specificity. To increase the sensitivity of hybridization assays, methods have been developed in which detection of the binding of the probe to its specific target is enhanced. For example, one commercially available kit uses genotype-specific RNA probes in either a high-risk or low-risk cocktail to detect the human papillomavirus (HPV) DNA in clinical specimens (see Chapter 66). Essentially, sensitivity of HPV detection by hybridization is increased by multimeric layering of reporter molecules, increasing their number on an antibody directed toward DNA-RNA hybrids using chemiluminescence; thus, sensitivity of detection is enhanced by virtue of greater signal produced (i.e., chemiluminescence) for each antibody bound to target. For PCR, nucleic acid is first extracted (released) from the organism or a clinical sample potentially containing the target organism by heat, chemical, or enzymatic methods. Numerous manual methods are available to accomplish this task, including a variety of commercially available kits that extract either RNA or DNA, depending on the specific target of interest. Other commercially available kits are designed to extract nucleic acids from specific types of clinical specimens, such as blood or tissues. Most recently, automated instruments (e.g., the Roche MagNaPure) have been introduced to extract nucleic acid from various sources, such as bacteria, viruses, tissue, and blood (Figure 8-7). Once extracted, target nucleic acid is added to the reaction mix containing all the necessary components for PCR (primers, nucleotides, covalent ions, buffer, and enzyme) and placed into a thermal cycler to undergo amplification (Figure 8-8). For PCR to begin, target nucleic acid must be in the single-stranded conformation for the second reaction, primer annealing, to occur. Denaturation to a single strand, which is not necessary for RNA targets, is accomplished by heating to 94°C (Figure 8-9). Of note, for many PCR procedures, especially those involving commonly encountered bacterial pathogens, disruption of the organism to release DNA is done in one step by heating the sample to 94°C. Primers are designed in pairs that flank the target sequence of interest (see Figure 8-9). When the primer pair is mixed with the denatured target DNA, one primer anneals to a specific site at one end of the target sequence of one target strand, and the other primer anneals to a specific site at the opposite end of the other, complementary target strand. Usually primers are designed to amplify an internal target nucleic acid sequence of 50 to 1000 base pairs. The annealing process is conducted at 50° to 58°C or higher. Annealing or hybridization of primers is optimized according to the nucleic acid sequence. The nucleic acid sequence of the primer determines the optimal annealing melting temperature (Tm) for the primers. The melting temperature is defined as the temperature at which 50% of the primers are hybridized to the appropriate complementary sequence. Because of the complementary binding of nucleotides, the melting temperature may be determined for a known nucleotide sequence. The melting temperature is calculated according to a simple formula: Annealing of primers to target sequences provides the necessary template format that allows the DNA polymerase to add nucleotides to the 3’ terminus (end) of each primer and extend sequence complementary to the target template (see Figure 8-9). Taq polymerase is the enzyme commonly used for primer extension, which occurs at 72°C. This enzyme is used because of its ability to function efficiently at elevated temperatures and to withstand the denaturing temperature of 94°C through several cycles. The ability to allow primer annealing and extension to occur at elevated temperatures without detriment to the polymerase increases the stringency of the reaction, thus decreasing the chance for amplification of nontarget nucleic acid (i.e., nonspecific amplification). As shown in Figure 8-9, for each target sequence originally present in the PCR mixture, two double-stranded fragments containing the target sequence are produced after one cycle. At the beginning of the second cycle of PCR, denaturation produces four templates to which the primers will anneal. After extension at the end of the second cycle, there will be four double-stranded fragments containing target nucleic acid. Therefore, with completion of each cycle, there is a doubling or logarithmic increase of target nucleic acid, and after the completion of 30 to 40 cycles, 107 to 108 target copies will be present in the reaction mixture. When the reliability of PCR for a particular amplicon has been well established, hybridization-based detection may not be necessary; confirming the presence of the correct-size amplicon may be sufficient. This is commonly accomplished by subjecting a portion of the PCR mixture, after amplification, to gel electrophoresis. After electrophoresis, the gel is stained with ethidium bromide to visualize the amplicon and, using molecular weight–size markers, the presence of amplicons of appropriate size (the size of the target sequence amplified depends on the primers selected for PCR) is confirmed (Figure 8-10). Multiplex PCR is a method by which more than one primer pair is included in the PCR mixture. This approach offers a couple of notable advantages. First, strategies including internal controls for PCR have been developed. For example, one primer pair can be directed at sequences present in all clinically relevant bacteria (i.e., the control or universal primers), and the second primer pair can be directed at a sequence specific for the particular gene of interest (i.e., the test primers). The control amplicon should always be detectable after PCR; absence of the internal control indicates that PCR conditions were not met, and the test must be repeated. When the control amplicon is detected, absence of the test amplicon can be more confidently interpreted to indicate the absence of target nucleic acid in the specimen rather than a failure of the PCR assay (Figure 8-11).
Nucleic Acid–Based Analytic Methods for Microbial Identification and Characterization
Overview of Molecular Methods
Nucleic Acid Hybridization Methods
Hybridization Steps and Components
Production and Labeling of Probe Nucleic Acid.
Mixture and Hybridization of Target and Probe.
Detection of Hybridization.
Hybridization Formats
Solution Format.
Solid Support Format.
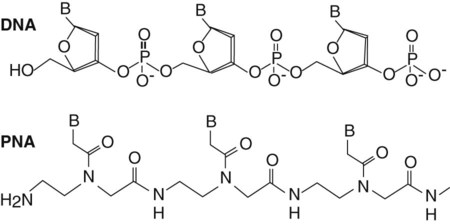
Peptide Nucleic Acid (PNA) Probes.
Hybridization with Signal Amplification.
Amplification Methods—PCR Based
Overview of PCR and Derivations
Extraction and Denaturation of Target Nucleic Acid.

Primer Annealing.
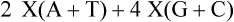
Extension of Primer-Target Duplex.
Detection of PCR Products.
Derivations of the PCR Method.
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree