The preparation of routine sections of bone after embedding requires the prior removal of calcium from the bone. As occasionally an unexpected underlying neoplastic process may be found, we routinely ensure that at least some of the tissue is decalcified in gentle decalcifying agents (ethylenediaminetetraacetic acid [EDTA] or other more recently available commercial products), as this allows molecular techniques to be undertaken when necessary. Calcium can be removed successfully and relatively quickly if some simple guidelines are followed. First, before decalcification, the tissue must be well fixed. Second, slices, not whole specimens, should be decalcified. Third, it must be realized that as the calcium is removed by acid, the acid is neutralized and depleted. Therefore, it is necessary that an adequate amount of acid be used, that the acid solution be changed frequently, and that a shaker be used to ensure that the acid thoroughly infiltrates the specimen. Following decalcification, all of the acid must be washed out of the specimen; otherwise, poor staining may result. Washing for at least 12 hours in running water usually ensures good differentiation of the hematoxylin and eosin stain.
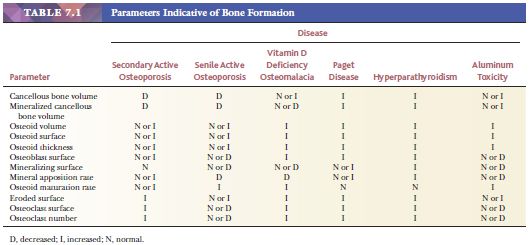
Histomorphometry, which is the quantification of morphologic features at the tissue and cellular levels, can further define the metabolic diseases of bone (6). The parameters indicative of bone formation include the percentages of active osteoblastic surface, osteoid surface, and mineralizing surface. Those indicative of bone mineralization include osteoid volume and mineral apposition rate. Those indicative of bone resorption include total eroded surface and active osteoclastic surface. Table 7.1 summarizes these parameters in the various metabolic bone diseases. Pathologists often say, “I don’t need a number to make the diagnosis of osteopenia.” This is true, but morphometry helps clarify the dynamics of the disease and subtle alterations in the balance between osteoblastic formative activity and osteoclastic resorption. Morphometry enhances the assessment of metabolic bone disease by supplementing the information gained from qualitative visual examination with quantitative information. Furthermore, the discipline required to make objective measurements can only improve visual diagnostic acumen.
METABOLIC DISEASES
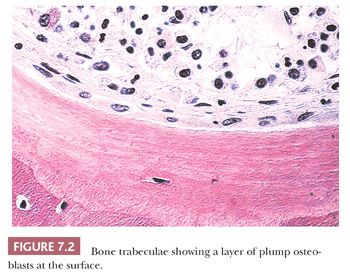
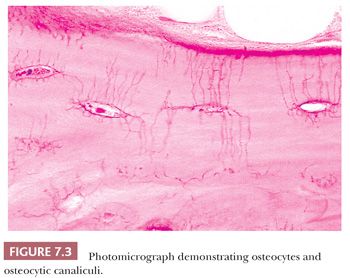
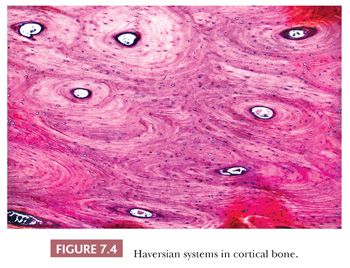
The quality and quantity of the extracellular matrix of the bone reflect cellular activity (7–9). Osteoblasts synthesize the collagenous matrix of the bone and regulate its mineralization. Osteoclasts, which may be multinucleated, are fewer in number than osteoblasts and are responsible for removing bone. For the amount of bone in the skeleton to remain stable, the rate of production must equal the rate of resorption; this equilibrium is often referred to as cell coupling or linkage. The rate of matrix production is reflected in the cytologic appearance of the osteoblasts, which range from flat (inactive) to plump (active) (Fig. 7.2). As bone formation proceeds, some of the osteoblasts are buried within the matrix and become osteocytes. The osteocytes maintain contact with each other and with the surface osteoblasts through cellular processes that run through a network of canaliculi within the bone matrix to form a functional syncytium, which is thought to play a major role in both mineral and structural homeostasis (Fig. 7.3). Cortical bone in adults is composed of closely applied, densely compact cylindrical units called osteons (Fig. 7.4). Primary osteons are formed in juveniles by the ingrowth of periosteal blood vessels that follow a “cutting cone” of osteoclasts, which tunnel through the existing cortex deposited by the periosteum. The tunnel, or haversian canal, formed by the osteoclasts and containing the vessels is subsequently partially filled in by osteoblasts, which deposit concentric layers of bone matrix. Secondary and subsequent osteons are formed during the process of remodeling by the outgrowth from existing haversian systems of new vessels; these are also preceded by a cluster of osteoclasts. Cortical bone has a predominantly structural role, whereas cancellous bone prevails in day to day metabolic activity. Osteoblastic activity is probably positively influenced by increased physical activity, parathyroid hormone, and certain growth factors. Activity of the osteoblasts is suppressed by inactivity and steroid hormones (10–12). Throughout life, maintenance of normal healthy bone is dependent on adequate load.
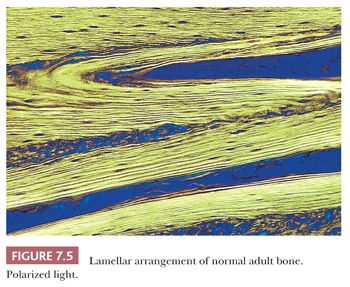
When the osteoblasts synthesize collagen, it is deposited in parallel layers to produce the lamellar pattern that characterizes normal adult bone (Fig. 7.5). Time is required to achieve this high degree of organization, and when bone is formed and deposited rapidly, as in the fetus, in fracture callus, or in certain neoplastic and hypermetabolic states, it is less obviously organized, and the collagen has a basket-like weave. This type of bone is referred to as woven or immature bone (Fig. 7.6). This is always abnormal after the age of 3 years and should alert the pathologist to look for underlying pathology. (The microscopic examination of collagen is greatly aided by the use of polarized light, which demonstrates not only the collagen fibers but also their organization.)

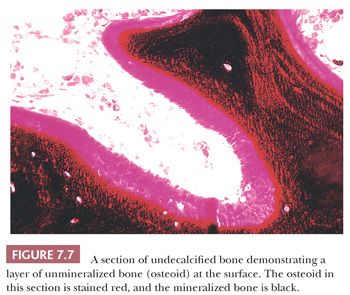
The distribution of calcium in bone cannot be studied by examining decalcified sections. However, it is possible to study the mineralization of the matrix if undecalcified sections are prepared. For the study of those diseases in which disturbed mineralization is suspected, this examination is essential (13). When the organic matrix is first deposited by the osteoblast, it is not mineralized; therefore, a layer of unmineralized matrix, termed osteoid, is always present at the formative surface of bone (Fig. 7.7). Normally, this layer is very thin because the time between matrix deposition and subsequent mineralization is short; however, when the rates of deposition and mineralization are unbalanced, the amount and extent of the osteoid increase, and hyperosteoidosis may develop. Hyperosteoidosis occurs basically for three reasons. First, in any condition in which bone formation is increased, the osteoid surfaces are both more extensive and thicker (e.g., fracture callus, Paget disease, hyperparathyroidism). Second, the mineralization of bone depends on the presence of many substances, including calcium, phosphorus, vitamin D, and alkaline phosphatase. If the amount of any of these substances is inadequate, mineralization is delayed, perhaps markedly. Third, the presence in bone of certain inhibitory or toxic substances, such as aluminum, iron, and fluoride, may block mineralization, even though the amounts of calcium, alkaline phosphatase, and vitamin D in the tissues are adequate.
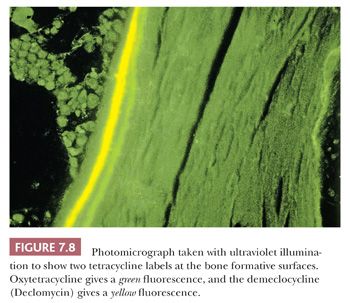
Tetracycline binds to actively mineralizing surfaces and exhibits fluorescence in ultraviolet light on microscopic examination; these features have made it a valuable aid in the study of bone and bone diseases (14). Pulsed doses of tetracycline can be administered to a patient before biopsy to determine the extent of mineralization and the amount of bone formed over a given time (Fig. 7.8). Additionally, the morphology of the tetracycline labels reflects the cause of the disease process. When mineralization is blocked, especially in an aluminum-associated disease, the label is not taken up. On the other hand, in patients with vitamin D deficiency, the labels are often extensive and smudged, whereas they are sharp in persons with normal bone formation (Fig. 7.9). (It should be noted that tetracycline may be passively absorbed onto a recently resorbed surface and thus confuse interpretation.)

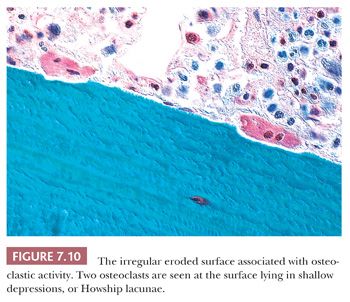
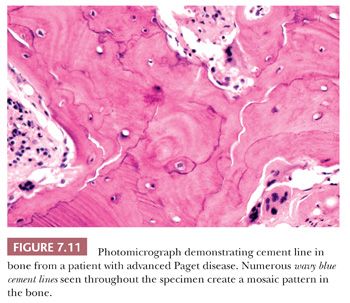
The resorption of bone by osteoclasts, rather than reflecting physiologic calcium homeostasis, is primarily associated with maintenance of the structural integrity of the skeleton. Morphologically, osteoclasts, which are large cells containing approximately two to four nuclei, adhere to the bone surface and are seen in depressions referred to as Howship lacunae or resorption bays (Fig. 7.10). In cancellous bone, resorption is generally limited to the bone surface, and the portions of the surface that have been resorbed can be recognized by an irregular, scalloped appearance, which contrasts with the smooth surface of formative or resting bone. Eventually, the resorptive surfaces are again covered by osteoblasts, and new bone forms on them. The junction between the original resorbed surface and the new bone is marked on sections stained with hematoxylin and eosin by a sharp basophilic line referred to as a reversal front or cement line (Fig. 7.11) (15).
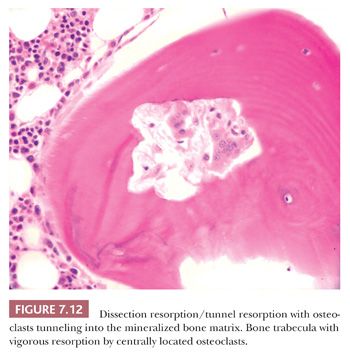
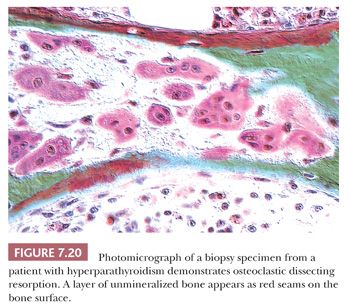
In diseases characterized by increased remodeling, such as Paget disease and osteoporosis, the osteoclastic activity, although often markedly increased, is generally confined to the surface. However, in hyperparathyroidism, whether primary or secondary, the osteoclasts characteristically tunnel into the mineralized bone matrix, creating a “wormhole” appearance referred to as dissecting resorption/tunnel resorption (Fig. 7.12). However, regardless of the cause or patterns of resorption, the architecture of the bone is distorted in all pathologic resorptive states.
ABNORMAL FORMATION OF THE ORGANIC MATRIX
The synthesis of collagen by the cells may be abnormal because of either genetic or acquired conditions. Examples of the former are osteogenesis imperfecta and Ehlers-Danlos syndrome, whereas examples of the latter are scurvy and lathyrism (a disease related to ingestion of certain legumes). Today, the condition most likely to be seen by the surgical pathologist is osteogenesis imperfecta.
Osteogenesis Imperfecta
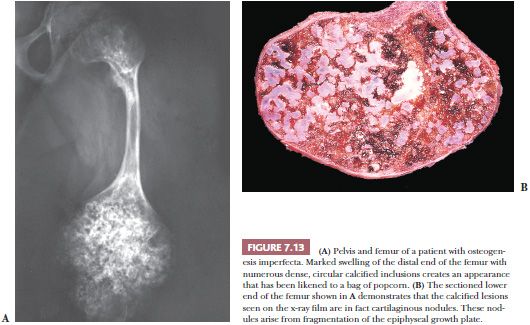
Osteogenesis imperfecta, one of the most common of the congenital connective tissue matrix diseases, comprises several distinct syndromes, some inherited as an autosomal dominant trait and others as a recessive trait; still others occur as spontaneous mutations. All have collagen mutations, mostly of COL1A1 and COL1A2, resulting in diminished quantity and quality of collagen type I (16). Persons with these various syndromes have in common a short stature and a propensity for fracture. Many of the patients also have poorly formed dentin, hearing loss, and frequently blue sclerae. Most of their clinical problems are caused by fractures, and those more severely affected, during the course of their childhood, sustain hundreds of major and minor fractures. The fractures occur more often in the lower limbs and, in a significant number of cases, involve the growth plates around the knee joints, giving rise eventually to growth plate fragmentation. Subsequent independent growth of the cartilage fragments causes swelling of the epiphyseal end of the bone. The deformed bone end filled with nodules of cartilage has been likened, on x-ray films, to a bag of popcorn (Fig. 7.13). These disruptions of the growth plate result in disproportionate shortening of the lower limbs in affected persons (17).
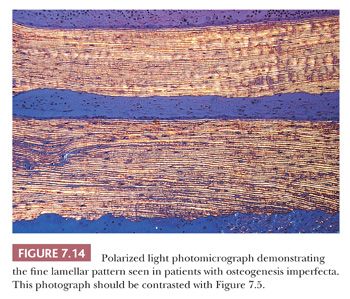
Bone samples from patients with the severe congenital form of osteogenesis imperfecta characteristically lack an organized trabecular pattern. The osteocytes are crowded within the bone, reflecting diminished collagen synthesis by the crowded osteoblasts at the surface. Frequently, large areas of woven bone are seen. In less severely affected persons, the bone is generally lamellar in pattern, although even in this bone, the osteocytes are crowded and the lamellae may be thinner than those in age-matched controls (Fig. 7.14). The lack of collagen synthesis by osteoblasts results in severely osteoporotic bone, which in turn leads to the numerous fractures characteristic of this condition.
ABNORMAL MINERALIZATION
The mineralization of bone depends on several factors, including vitamin D, calcium, phosphate, and alkaline phosphatase; it may be disturbed by the presence of certain metals, such as lead, strontium, iron, and aluminum.
Because routine microscopic examination of bone requires decalcification of the tissue before embedding and sectioning, disease resulting from abnormal mineralization is likely to be overlooked by pathologists unless the clinician has alerted them to the possibility. Plastic embedding can be used to prepare undecalcified sections. If small pieces of only cancellous bone are used, even paraffin-embedded undecalcified tissue may provide adequate sections for diagnosis.
Osteomalacia and Rickets
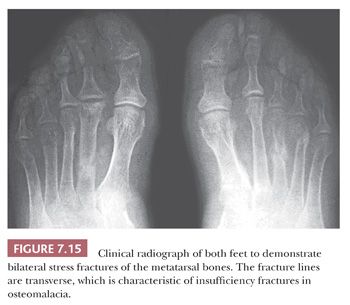
In general, the terms osteomalacia (in adults) and rickets (in children) are used to describe those diseases that result from a deficiency in vitamin D, an abnormality in the metabolism of vitamin D, or a deficiency of calcium in the diet. The most common symptom of osteomalacia is bone pain, which is usually generalized and often vague. In addition, the low calcium level may cause muscle weakness, which is often profound (18). Radiographic examination reveals generalized osteopenia with the classic finding of multiple bilateral and symmetric partial linear fractures of the bone, commonly referred to as insufficiency fractures (Fig. 7.15).
On microscopic examination, the most striking abnormalities are a massive increase in the amount of unmineralized bone (up to 40% or 50% of the total bone volume) and disorganization of the trabecular architecture. The mineralization front, which is the junction between the osteoid and mineralized bone, is very irregular, granular, and fuzzy. In addition to an increase in osteoid volume, bone volume is often increased overall as a consequence of increased osteoblastic activity (Fig. 7.16).

Rickets is the childhood manifestation of osteomalacia. In childhood, the anatomic changes are found most characteristically around the metaphyses of the most rapidly growing bones—that is, around the knee and wrist joints. On x-ray films, the epiphyseal growth plates are irregular and broadened and have a characteristic cup shape. Microscopically, the growth plate is thickened and poorly defined, especially on its metaphyseal side, where tongues of uncalcified cartilage can be seen extending into the metaphysis. As in adults, the bone shows extensive, wide osteoid seams.
In addition to the classic forms of osteomalacia and rickets secondary to calcium and vitamin D deficiencies, severe osteomalacia may occasionally be secondary to hypophosphatemia. Usually, the hypophosphatemia is the result of increased urinary phosphate loss, which may be the consequence of a primary renal tubular defect, diuretic therapy, or hyperparathyroidism. Very rarely, hypophosphatemia is associated with FGF23-producing, phosphaturic mesenchymal tumor, which has been documented in many locations and may be very tiny, resulting in oncogenic osteomalacia. In this instance, resection of the tumor generally results in complete resolution of the osteomalacia. Therefore, it is important to look for an occult tumor in cases of osteomalacia without a clear cause (19–21).
Hypophosphatasia
Hypophosphatasia (not to be confused with hypophosphatemia) is a rare genetic disease characterized by a disturbance in the synthesis of the enzyme alkaline phosphatase (22). It takes two forms. The first is inherited as an autosomal recessive trait that manifests as severe disease in infants. In general, when hypophosphatasia is diagnosed in infants younger than 6 months of age, it follows a rapidly progressive fatal course. The second form is an autosomal dominant condition that may not become evident until adulthood. In these cases, the disease is less severe and often asymptomatic.
The disorder is characterized clinically by decreased levels of alkaline phosphatase in the blood, bone, intestines, liver, and kidneys. The serum phosphorus and calcium levels are usually normal. In less severe cases, hypophosphatasia may not present clinically until the fourth, fifth, or sixth decade of life. Patients often have a childhood history of a rickets-like disorder, short stature, and deformed extremities.
Microscopic examination of the tissue from affected infants reveals increased osteoid and irregular epiphyseal cartilage with lengthened chondrocyte columns. Histopathologic examination of bone from adult patients reveals an osteomalacic picture, with increased quantities of unmineralized bone. Unlike the osteomalacia of vitamin D deficiency, that of hypophosphatasia is characterized by a paucity of osteoblasts.
Metal Toxicity
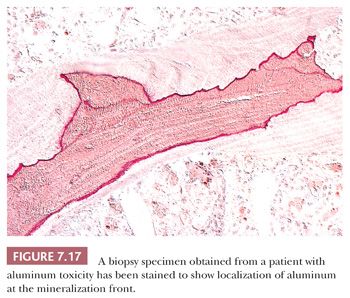
After the introduction of renal dialysis, many patients undergoing this procedure were treated with large doses of antacids to bind dietary phosphate and thereby prevent hyperphosphatemia. Eventually, it became apparent that the aluminum in the antacid had been incorporated into the bone and other body tissues of these patients. Several disease states resulted, most importantly aluminum-induced encephalopathy and aluminum-induced bone disease (23). In aluminum-induced bone disease, the amount of osteoid in the skeletal tissues may be considerably elevated; however, in this form of hyperosteoidosis, unlike that of vitamin D deficiency, the mineralization front is well demarcated, and the osteoblastic activity is minimal. Aurintricarboxylic acid stain can be used to demonstrate aluminum along the mineralization front (Fig. 7.17).
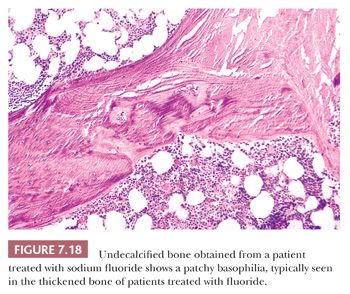
To a much lesser extent, both iron (24) and fluoride interfere with the deposition of calcium within bone. In conditions characterized by high levels of iron (e.g., thalassemia) or fluoride (e.g., fluorosis), osteoid on the surface of the bone is increased (Fig. 7.18).
ABNORMAL RESORPTION
Several conditions are characterized by increased resorption of bone, including hyperparathyroidism, Paget disease, and osteoporosis. Osteopetrosis is also thought to be a disease related to abnormal resorption; although osteoclasts are frequently abundant, they appear not to resorb the calcified cartilage in bone formed by endochondral ossification.
Hyperparathyroidism
The overproduction of parathyroid hormone may be either a primary or a secondary phenomenon. The most common cause of primary hyperparathyroidism is an adenoma of one of the parathyroid glands; more rarely, it is a carcinoma or hyperplasia of obscure origin. Hyperparathyroidism is characterized biochemically by marked hypercalcemia and hypophosphatemia. The patients are usually young to middle-aged adults who currently are most often identified by hypercalcemia on routine blood testing. Some present with a history of recurrent kidney stones, peptic ulcer, or less specific complaints, such as nausea, vomiting, weakness, and headaches (25). Although kidney disease is the most common clinical presentation, bone disease is present in approximately one-fourth of patients in the form of bone pain or, rarely, pathologic fracture.
Secondary hyperparathyroidism is generally the result of chronic renal failure in which, in association with phosphate retention, hypocalcemia develops. This process initiates compensatory or secondary hyperparathyroidism (26).
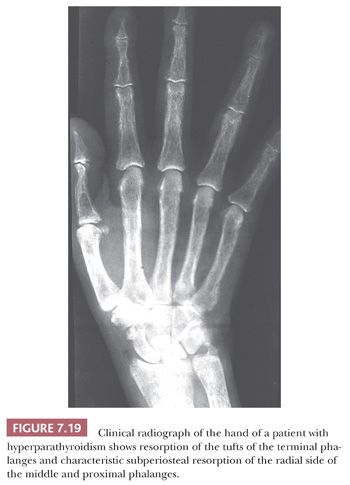
Whether the disease is primary or secondary, the radiologic and pathologic features found in the bone are similar (27). Roentgenographic examination may reveal diffuse osteopenia, but the most characteristic findings are seen on roentgenograms of the hand, where erosion of the tufts of the phalanges and subperiosteal cortical resorption, especially on the radial side of the phalanges, are apparent (Fig. 7.19). Very rarely, large lytic lesions mimicking bone tumors may develop within the skeleton. These lesions, known as brown tumors because of their color, may be solitary or multiple, and when multiple, they give rise to the classic presentation described by von Recklinghausen—osteitis fibrosa cystica. When solitary, they may be mistaken radiologically and histologically for an aneurysmal bone cyst or a giant cell tumor. Thus, whenever diagnosis of a giant cell–containing lesion of bone is made, the serum calcium and parathyroid hormone levels should be checked to exclude a brown tumor.
On microscopic examination, the characteristic finding in hyperparathyroidism is increased osteoclastic activity; burrowing of the osteoclasts into the bone matrix creates a tunneled appearance (Fig. 7.20). In addition to increased resorption, bone formation is markedly increased; this is often associated with peritrabecular fibrosis (Fig. 7.21). On occasion, the peritrabecular fibrosis must be distinguished from the changes associated with myelofibrosis. Whereas in myelofibrosis the increased fibrous tissue is generally distributed diffusely throughout the bone marrow, in hyperparathyroidism, the fibrosis hugs the trabecular surface. Another differential diagnostic consideration is the acute phase of Paget disease. In Paget disease, the osteoclasts are generally at the surface of the bone trabeculae, and they do not form tunnels. Furthermore, the osteoclasts of Paget disease frequently contain many more nuclei than the osteoclasts of hyperparathyroidism. Nevertheless, because it is not always possible to distinguish the two conditions microscopically, the clinical presentation and the biochemical findings are essential for making the correct diagnosis.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


