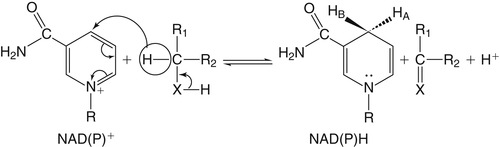
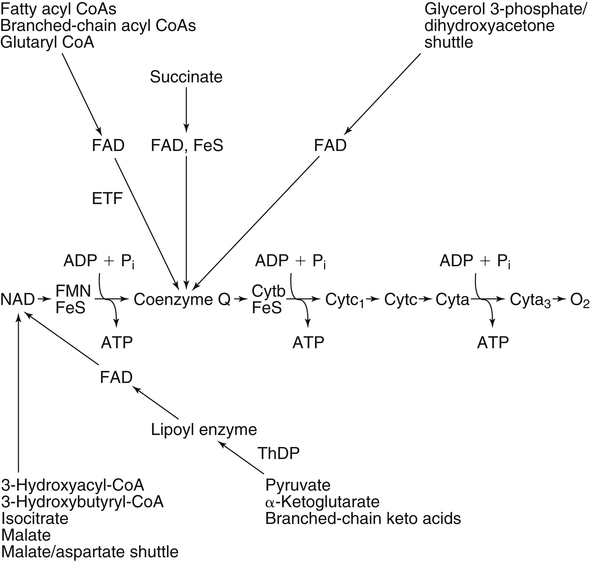
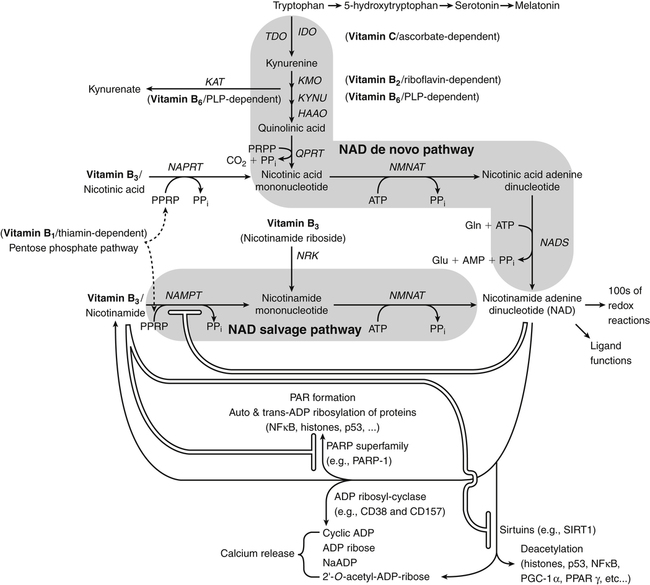
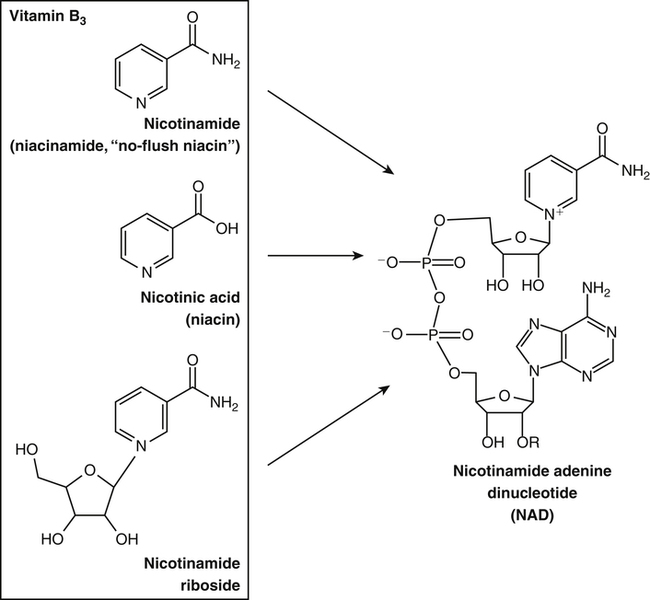
By the 1950s medical doctors who witnessed pellagra noticed its similarities to schizophrenia. These doctors theorized that schizophrenics might have a genetic disorder that required far greater levels of dietary niacin. Experiments revealed that fixed administration of gram quantities of niacin to schizophrenic patients frequently resulted in therapeutic benefit with minimal or no adversity (Hoffer et al., 1957). However, this approach remains controversial and requires additional study. As a result of these initial experiments focused on schizophrenia, it was also determined that high-dose niacin (as nicotinic acid) is an effective treatment for dyslipidemia. Today high-dose niacin is a more effective elevator of high-density lipoprotein (“good” cholesterol) than any other pharmaceutical. This approach simultaneously lowers cholesterol, triacylglycerols, and very-low-density lipoprotein. Administration of high doses of various NAD precursors remains an active area of research, especially based on the many favorable results obtained in animal models of human diseases. Vitamin B3 is defined as the precursor to NAD and potentially includes three different molecular forms: nicotinic acid, niacinamide, and nicotinamide riboside. Nicotinic acid or pyridine-3-carboxylic acid is sometimes referred to as “flushing” niacin, whereas niacinamide or pyridine-3-carboxamide is referred to as “flush-free” niacin. The third form of vitamin B3, nicotinamide riboside, was recently discovered in 2004 and is not used clinically yet (Bieganowski and Brenner, 2004). Structures for the three molecules are shown in Figure 24-1. Niacin-responsive genetic diseases such as aldehyde dehydrogenase and glucose-6-phosphate 1-dehydrogenase can be rescued with high doses of niacin (Table 24-1). High-dose vitamin therapies stimulate variant enzymes with decreased coenzyme binding affinity (increased Km), thereby improving the metabolic dysfunction (Ames et al., 2002). Given that the number of proteins that require NAD is greater than that for any other cofactor, scores of unmapped genetic polymorphisms are predicted to be responsive to high doses of NAD precursors, many of which have yet to be examined. TABLE 24-1 Niacin-Responsive Genetic Disorders aldehydes to acids, and certain amino acids to keto acids. The common mechanism of operation for NAD(P[H]) cofactor redox reactions is generalized in Figure 24-2. Most dehydrogenases that use NAD or NADP function reversibly. Generally enzymes use NAD(H) in catabolic reactions, whereas NADP(H) is more commonly involved in anabolic (biosynthetic) reactions. For example, NADPH serves as an important reducing agent for the synthesis of fats and steroids (e.g., reactions catalyzed by 3-ketoacyl reductase, enoyl reductase, and 3-hydroxy-3-methylglutaryl-coenzyme A [HMG-CoA] reductase). Both NAD and NADP are part of the intracellular respiratory mechanism of all cells. They assist in the stepwise transfer of electrons or reducing equivalents from various energy substrates to the cytochromes. NADH usually donates its electrons to a flavin coenzyme in the mitochondrial electron transport chain responsible for ATP production. These reactions are outlined in Figure 24-3. A major source of NADH in the mitochondria is the β-oxidation of fatty acids. The main extramitochondrial source is NADH formed in glycolysis. Reducing equivalents are carried to the mitochondria by the malate–aspartate or the glycerol phosphate–dihydroxyacetone shuttles (see Chapter 12). The approximately 57 different human cytochrome P450 monooxygenase systems use reducing equivalents of NADH and NADPH for a wide variety of functions, including synthesis and degradation of drugs, xenobiotics, prostaglandins, leukotrienes, retinoic acid, vitamin D, cholesterol, bile, and steroids (Nebert and Russell, 2002). Four classes of enzymes play dominant roles in controlling NAD levels in response to DNA damage, immune activation, and other stimuli. These enzymes are poly(ADP-ribose) polymerase 1 (PARP1 through PARP18), the NAD-dependent deacetylases (sirtuin 1 through sirtuin 7), the ADP ribosyl-cyclases (CD38 and CD157), and indoleamine 2,3-dioxygenase (IDO). The first three of these enzymes produce nicotinamide as a side product that is recycled back to NAD via the salvage pathway as shown in Figure 24-4. Absorption of some nicotinic acid occurs by passive diffusion in the stomach. Both nicotinic acid and its amide are absorbed in the small intestine by a sodium (Na+)-dependent saturable process as well as by passive diffusion that increases at higher nonphysiological concentrations of the vitamin (Bechgaard and Jespersen, 1977). Coenzyme forms of niacin in the gastrointestinal tract are first rapidly hydrolyzed to nicotinamide mononucleotide (NMN) by nonspecific pyrophosphatases in the intestinal lumen (Gross and Henderson, 1983). Alkaline phosphatase catalyzes further cleavage to nicotinamide riboside, which is converted to NAD by a two-step pathway starting with a reaction catalyzed by nicotinamide riboside kinase (see Figure 24-4) (Bieganowski and Brenner, 2004). NAD glycohydrolases (NADases) within mucosal cells may contribute to the breakdown of the coenzymes to nicotinamide. Once in the plasma, niacin enters cells by active or passive diffusion followed by metabolic trapping. Active diffusion of niacin into erythrocytes involves an anion transporter protein. Both erythrocytes and liver rapidly remove and convert niacin to NAD. Niacin and Cancer 1. Given that cancer is a disease defined by genetic mutation and rapid cellular proliferation, should niacin intake be restricted during chemotherapy? 2. Caloric restriction also delays the onset of cancer. The body normally undergoes the process of cachexia during cancer. Given that cachexia resembles pellagra, which involves vitamin depletion, and that inhibition of NAD biosynthesis is more likely to kill rapidly growing cancer cells rather than healthy cells, is it possible that cachexia may be in part a desirable response of the body to attempt to control cancer? Data from U.S. Department of Agriculture (USDA), Agricultural Research Service (ARS). (2010). USDA National Nutrient Database for Standard Reference, Release 23. Retrieved from www.ars.usda.gov/ba/bhnrc/ndl riboflavin, pyridoxyl phosphate (vitamin B6), and ascorbate (vitamin C) (see Figure 24-4). The de novo pathway is highly regulated by the immune system in specific cell types. Interferon gamma activates the rate-limiting enzyme, IDO, which is required for interferon gamma’s biological activities. Many intermediates in this NAD synthetic pathway such as kynurenine and kynurenate have significant physiological roles. Persistent activation of IDO is seen in many autoimmune diseases and in cancer, where decreased serum tryptophan is a common diagnostic indicator of poor prognosis. The complementary administration of high doses of alternative NAD precursors has been repeatedly shown to rescue these pathogenic processes in both animal models and some clinical cases (Penberthy, 2007). The NAD salvage pathway recycles nicotinamide produced as a side product of NAD-degradation by PARP, sirtuin, and ADP ribosyl-cyclases back to NAD (see Figure 24-4). Nicotinamide phosphoribosyltransferase (NAMPT) is the rate-limiting enzyme controlling the rate of recycling of nicotinamide to NAD (Revollo et al., 2004). Importantly, NAMPT gene expression is strongly induced in response to a wide variety of stresses to provide an adequate level of NAD. Increased nicotinamide mononucleotide adenylyltransferse (NMNAT)-1, 2, or 3 also provides tremendous cell survival benefit in a wide range of stresses, particularly as seen in neurodegenerative models (Sasaki et al., 2006). Although NAMPT may be rate limiting for the salvage pathway under basal conditions, the NMNAT enzymes may limit NAD synthesis under stress-induced conditions where the levels of NAMPT have been dramatically increased. NAD inhibits NAMPT activity but not nicotinate phosphoribosyltransferase (NAPRT). This is partially why nicotinic acid provides greater levels of intracellular NAD than nicotinamide for many cell types. Over the past several decades, dyslipidemia has been commonly treated with 3 to 5 g of nicotinic acid daily without serious adverse events (Carlson, 2005; Guyton and Bays, 2007). However, the flush response can be exceedingly unpleasant. The upper tolerable intake level (UL) for adults is designated at 35 mg/day of niacin, limited to niacin Case Study of Pellagra in an Adult Woman Oakley and Wallace (1994) reported a case involving a young woman in New Zealand who had a variety of neurological and dermal symptoms consistent with pellagra. However, she did not have diarrhea. Her symptoms were precipitated by prolonged lactation and increased activity. She had breast-fed her daughter for 3 years and had been particularly active while building her own home. Dietary intakes of niacin equivalents were within normal range. Her daily total excretion of urinary amino acids was abnormally high (77 mmol/L). Chromatography of urinary amino acids revealed elevated loss of tryptophan and other large neutral amino acids (alanine, serine, threonine, valine, methionine, isoleucine, tyrosine, and phenylalanine), whereas taurine, glycine, ornithine, lysine, arginine, and aspartate were present in normal amounts. Her symptoms were resolved after taking nicotinamide plus other B vitamins. 1. From the information given, what underlying metabolic abnormality was probably responsible for the development of pellagra in this woman with normal intake of niacin? 2. The first symptoms of pellagra secondary to inherited disorders usually occur in childhood. Why would periods of rapid growth or of prolonged lactation as in this case precipitate the appearance of pellagra? Why might increased activity precipitate the appearance of pellagra? obtained from synthetic sources or fortified foods, based on the dose of nicotinic acid that can be associated with flushing effects. Although the flush can be uncomfortable, it is in fact linked to the beneficial effect of correcting dyslipidemia, because nicotinamide does not exert these therapeutically beneficial effects on lipid profiles. Biochemical assessment of niacin nutritional status is usually based on whole blood measures of NAD/NADP (Jacobson and Jacobson, 1997). Restricting niacin/tryptophan intake to 50% of the Recommended Dietary Allowance (RDA) decreases NAD by 70% within 5 weeks while NADP levels remain constant. A decrease in NAD levels precedes pellagra symptoms and are frequently seen in carcinoid syndrome patients.
Niacin, Riboflavin, and Thiamin
Niacin History
Niacin Nomenclature, Structure, and Biochemistry
Niacin Physiological Function
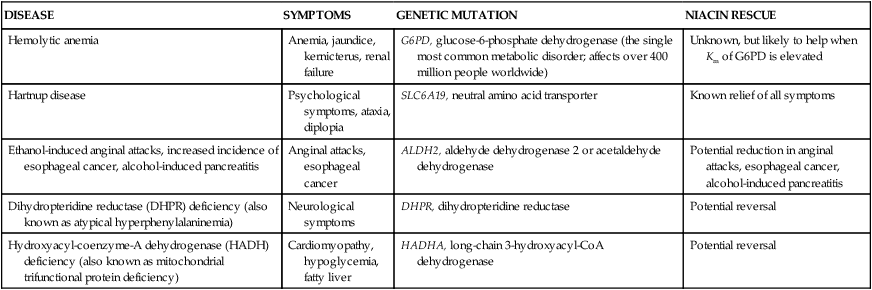
Niacin-Responsive Genetic Disorders
DISEASE
SYMPTOMS
GENETIC MUTATION
NIACIN RESCUE
Hemolytic anemia
Anemia, jaundice, kernicterus, renal failure
G6PD, glucose-6-phosphate dehydrogenase (the single most common metabolic disorder; affects over 400 million people worldwide)
Unknown, but likely to help when Km of G6PD is elevated
Hartnup disease
Psychological symptoms, ataxia, diplopia
SLC6A19, neutral amino acid transporter
Known relief of all symptoms
Ethanol-induced anginal attacks, increased incidence of esophageal cancer, alcohol-induced pancreatitis
Anginal attacks, esophageal cancer
ALDH2, aldehyde dehydrogenase 2 or acetaldehyde dehydrogenase
Potential reduction in anginal attacks, esophageal cancer, alcohol-induced pancreatitis
Dihydropteridine reductase (DHPR) deficiency (also known as atypical hyperphenylalaninemia)
Neurological symptoms
DHPR, dihydropteridine reductase
Potential reversal
Hydroxyacyl-coenzyme-A dehydrogenase (HADH) deficiency (also known as mitochondrial trifunctional protein deficiency)
Cardiomyopathy, hypoglycemia, fatty liver
HADHA, long-chain 3-hydroxyacyl-CoA dehydrogenase
Potential reversal
Proteins That Require Niacin
Redox Reactions
Nonredox Reactions and Control of Nad Levels
Niacin Sources, Chemical Stability, and Admet
Sources
ADMET
Absorption
Distribution and Metabolism
 CLINICAL CORRELATION
CLINICAL CORRELATION
 FOOD SOURCES OF PREFORMED NIACIN
FOOD SOURCES OF PREFORMED NIACIN
Toxicity
 CLINICAL CORRELATION
CLINICAL CORRELATION
Biochemical Assessment of Niacin Nutriture, DIETARY Requirements, and High-Dose Responses
Biochemical Assessment of Niacin Nutriture
Dietary Requirements
 DRIs Across the Life Cycle: Niacin
DRIs Across the Life Cycle: Niacin
mg NE per Day
Infants
RDA
UL∗
0 through 6 mo
2 (AI)
ND
7 through 12 mo
4 (AI)
ND
Children
1 through 3 yr
6
10
4 through 8 yr
8
15
9 through 13 yr
12
20
Males
14 through 18 yr
16
30
≥19 yr
16
35
Females
14 through 18 yr
14
30
≥19 yr
14
35
Pregnant
<19 yr
18
30
≥19 yr
18
35
Lactating
<19 yr
17
30
≥19 yr
17
35 ![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


 NUTRITION INSIGHT
NUTRITION INSIGHT