Well-differentiated PENs can also be classified as functional or nonfunctional based on the presence or absence of an associated clinically recognizable syndrome (Table 56-2). These syndromes are the result of the secretion of biologically active hormones by the tumors and are confirmed by measurable elevations of the hormones in the blood. The most common functional PENs include insulinomas, gastrinomas, vasoactive intestinal polypeptide-omas (VIPomas), glucagonomas, and somatostatinomas. The incidence of these lesions ranges from 1 per 1 million for insulinomas to 1 per 40 million for somatostatinomas.24 Even less common PENs secreting calcitonin,25,26 parathyroid hormone–related protein,27 growth hormone–releasing factor, and adrenocorticotropic hormone28 have been reported. Nonfunctional PENs are classified as such due to their lack of an associated clinical syndrome. Some of the tumors in this group do secrete elevated amounts of hormones, including chromogranin A, which can be detected in either the serum or in surgical specimens using immunohistochemistry.29 These secreted hormones either produce no clinical syndromes, as is seen with tumors that secrete pancreatic polypeptide,30 or secrete hormones in subclinical amounts or inactive forms. Traditionally, functional PENs were reported to comprise the majority of PENs. As methods of detecting these lesions and patterns of presentation have evolved, due primarily to the widespread use of high-quality cross-sectional imaging, nonfunctional PENs now comprise the vast majority of surgically resected cases.18,31
The least common group of PENs is the poorly differentiated or high-grade endocrine carcinomas. These are aggressive tumors characterized by their high mitotic count and proliferation index (>20 mitotic figures per 10 high-powered fields, Ki-67 >20%).23,32 These tumors primarily occur in adults and have a male predominance. Some have been reported to be functional, producing varied clinical syndromes (commonly gastrinoma, VIPoma, glucagonoma, and, less frequently, insulinoma). Prognosis is often poor, with the clinical course varying from a rapid decline to a more indolent, prolonged survival.
Table 56-2 Classification of Functional Pancreatic Endocrine Tumors

MOLECULAR GENETICS
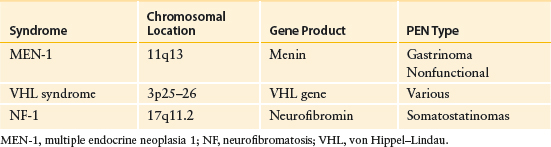
The majority of PENs are sporadic. Some of them, however, occur as part of inherited familial syndromes such as multiple endocrine neoplasia type 1 (MEN-1), von Hippel–Lindau (VHL) syndrome, neurofibromatosis (NF-1), and tuberous sclerosis (TSC) (see section below on genetic syndromes) (Table 56-3). Recent advances in high throughput DNA sequencing techniques have provided new insights into the genesis of PENs and possible reasons why certain tumors behave more aggressively than others as well as why tumors may respond more favorable to specific therapies (i.e., a more personalized approach to therapy of PENs).
Whole-Exome Sequencing of PENs
Specifically, in a 2011 landmark paper, Jiao et al. sequenced ∼18,000 coding genes of 10 clinically well-characterized PENs.33 Technically, this work dovetailed eloquently and aligned with the group’s previous work of sequencing and analyzing multiple pancreatic ductal adenocarcinoma genomes.34 Importantly, the investigators microdissected the samples in an effort to achieve a high purity and quality of DNA from neoplastic tissue. In the PENs cancer genomes compared to pancreatic ductal adenocarcinoma genomes a number of differences were discovered including >50% fewer mutations in PENs compared to pancreatic adenocarcinoma genomes; and commonly mutated genes such KRAS were not mutated in PENs (Table 56-4).35 These genetic data support the notion that the chromosomal instability (CIN) and tumorigenesis process between these two pathologically distant pancreatic neoplasms are initiated by unique molecular and/or environmental events. The study went on to validate the common findings in the 10 PENs (labeled a discovery set) with an additional 58 PENs. Validating previous work and also underscoring the importance of the disruption in the chromatin-remodeling pathway in PENS, a majority of PENs harbored a tumor-specific inactivating mutation in MEN-1. Additional genes functionally important in a chromatin-remodeling complex, DAXX (death-domain–associated protein) and ATRX (alpha thalassemia/mental retardation syndrome X-linked), were frequently mutated in PENs as well. Correlating the clinical outcomes of these tumors, the study demonstrated that patients in this cohort with MEN-1, DAXX, and ATRX mutated tumors had unique outcomes compared to the other PENs. More recent work published in 2014, correlated DAXX and ATRX mutations, activation of alternative lengthening of telomeres, and global chromosomal alterations with pathologic features and outcome data.36 Interestingly, this study identified that PENs harboring DAXX and ATRX mutations were more likely to have CIN and patients with this subtype of PENs had a reduction in survival.
Table 56-3 Familial Genetic Syndromes Associated with Pancreatic Endocrine Neoplasms (PENs)
This landmark sequencing work underscored the potential clinical importance of mutations in the mTOR pathway in a subset of PEN patients. As a reminder, the drug rapamycin targets mTOR, and thus, this finding should provide the framework in which we may stratify PEN patients for TOR inhibitor–based therapies (e.g., everolimus).33 As this work demonstrated that mTOR dysregulation may be an important predictive marker, others have shown that in a large panel of neuroendocrine tumors (195 of which only 19 where pancreatic) mTOR overexpression and/or its downstream-activated targets were associated with worse clinical outcomes (i.e., adverse prognostic markers).37 This work provides another instance where a poor prognostic marker (for even development of disease) may serve counterintuitively as a positive predictive marker (for everolimus); an established biomarker, BRCA2, acts in a similar fashion.
Table 56-4 Portal Vein Sampling

Genetic Links and Syndromes Related to PENs
Significant progress has been made in the genetic understanding of the MEN-1 syndrome in relation to PENs.38 Chromosomal linkage studies have localized the genetic defect to the 11q13 locus, and studies of DNA markers have localized the MEN-1 gene between PYGM and D11S97. The gene contains 10 exons that code for a 610-amino-acid protein called menin, whose function is unknown, although it is classically labeled as a tumor suppressor gene. Some studies provide a possible explanation for loss of this gene in neuroendocrine tumors.39 The menin protein is expressed in diverse tissues and is highly conserved evolutionarily. Menin is predominately a nuclear protein, which binds to JunD and may repress JunD-mediated transcription. Studies in patients with MEN-1 have shown allelic deletions at chromosome 11q13 in nearly 100% of parathyroid tumors, 85% of nongastrinoma islet cell tumors, and up to 40% of gastrinomas. In patients with sporadic tumors (without MEN-1), 11q13 deletions are seen in about 25%, 20%, and almost 50% of parathyroid tumors, nongastrinoma PENs, and gastrinomas, respectively. Recently it has been shown that a comprehensive genetic testing program for patients at risk for MEN-1 can identify patients harboring a MEN-1 mutation almost 10 years before the development of clinical signs or symptoms of disease.40 Since MEN-1 loss has been detected in both the sporadic and the familial forms of PENs, the menin pathway is most likely involved in the overall pathogenesis of this disease, whether familial or sporadic in nature.39
Less frequently than MEN-1, PENs may be associated with VHL syndrome. VHL syndrome is another autosomal dominant inheritance disease that includes many clinical disorders41 including retinal hemangioblastomas, cerebellar and medullary hemangioblastomas, and PENs. PENs are found in a small percentage of patients with VHL syndrome. A mutation in the VHL gene, a tumor suppressor located on chromosome 3p25–26, which regulates hypoxia-induced cell proliferation, is responsible. Although germline mutations with loss of heterozygosity (LOH) are associated with this disease, it has been proposed that other tumor suppressors most likely cooperate with VHL in order to form PENs. VHL-mutated PENs have been shown to have specific defects in angiogenesis and hypoxia-inducible factor pathways.42
NF-1 (von Recklinghausen disease) is an autosomal dominant disorder that produces a well-described clinical syndrome characterized by café-au-lait spots and neurofibromas. These patients may develop pancreatic somatostatinomas, often near the ampulla of Vater. The NF-1 gene is a tumor suppressor gene located on 17q11.2 that encodes for neurofibromin, a regulator of the mammalian target of rapamycin (mTOR) pathway. Loss of NF-1 results in mTOR activation and tumor development.43,44
Complementary Progression Modeling
Based on the data from the Marinoni study that attempted to molecularly subtype PENs,36 a stepwise progression model of PENs were put forth: (1) initiation occurs (unknown mechanism); (2) DAXX/ATRX mutations induce transformation; (3) alternative lengthening of telomere (ALT)45 activation and CIN; (4) clonal heterogeneity and a selection of clones; and (5) metastases.36 Complementary to this work, Zhang described a “double hit model” of PEN progression35 wherein a first mutational hit (e.g., MEN1 or p53) can induce cell cycle progression, even under harsh tumor microenvironment conditions (low glucose). Then, the second hit (ATRX and mTOR) can cause cell growth and enhance cell invasion and metastatic potential,35 even in the absence of cell–cell adhesion. These models highlight our recent and enhanced knowledge of the molecular etiology of PEN. However, future work will need to define the main initiating event that sets the stage for PEN transformation. Since KRAS activation appears to be unnecessary, other nonclassical genetic events (e.g., epigenetics and posttranscriptional gene regulation) should be surveyed as critical key events in this tumorigenesis process.
Molecular Targeting of PENs
In summary, PENs are becoming easier to characterize molecularly (and clinically) due to large scale sequencing efforts and a better understanding behind the molecular biologic aspects of these pancreatic tumors. Ongoing efforts to search for candidate genes and novel pathways that set the stage for CIN and initiation of tumorigenesis in these tumors will aid in unraveling the molecular etiology of this disease and perhaps even better druggable targets (see Table 56-4 for molecular pathways to target in PENs vs. pancreatic ductal adenocarcinoma). Still, the molecular characterization of these tumors have helped to pave the way for clinical trials targeting tyrosine kinases (e.g., sunitinib) and the mTOR pathways (e.g., everolimus). In fact, recent work explored targeting multiple points throughout the dysregulated pathway (PI3K/AKT/mTOR) in PENs by combining PI3K inhibitors with mTOR inhibitors and demonstrated that this combination may break initial and acquired drug resistance in PENs.46 Future molecular interrogation of PENs combined with these types of preclinical targeting approaches should continue to move the field closer to a personalized approach to treating PENs with better targeted therapies.
PRESENTATION AND EVALUATION
There are three primary ways by which patients with PENs come to clinical attention: the incidental discovery of a mass in the pancreas during cross-sectional imaging, symptoms secondary to the mass effect of a lesion in the pancreas (i.e., obstructive jaundice or pain), and, as a consequence of the symptoms of a syndrome associated with a functional PEN. As mentioned previously, incidentally detected nonfunctional PENs currently comprise the majority of clinically relevant tumors. They are typically hypervascular on imaging studies such as computed tomography (CT) or magnetic resonance imaging (MRI). In the absence of any clinical syndrome, these lesions can be managed as any other incidental pancreatic lesion. Typically, the size of a PEN at discovery guides decision making between three standard options: serial observation, endoscopic ultrasound-guided biopsy, or definitive surgical resection. Size greater than 2 cm is a standard stratification measure, with lesions larger than this usually managed in a more aggressive fashion. If a nonfunctional PEN is suspected, baseline serum levels of chromogranin A and pancreatic polypeptide can be useful diagnostic markers prior to surgical resection. Chromogranin A is a 49-kDa protein contained within the neurosecretory vesicles of PENs, whose levels should be measured prior to surgical resection and are expected to significantly decline in the postoperative period with removal of the tumor burden. The chromogranin A levels can be tracked over time to document tumor recurrence, often before the lesions become visible on cross-sectional imaging studies.47 Pancreatic polypeptide is secreted by the PP cells of the islets of Langerhans and can also be used to track patients with PENs, though its sensitivity (63%) is lower than that of chromogranin A.48 Neuron-specific enolase is another tumor marker that is elevated in approximately 50% of PENs, most commonly in patients with pulmonary metastases.49
PENs presenting due to mass effect on surrounding structures resulting in jaundice, pain, or gastric outlet obstruction are uncommon. These lesions should be addressed as any other symptomatic pancreatic lesion with definitive surgical resection if clinically appropriate.
Patients presenting with symptoms from a functional PEN can be a diagnostic challenge. Three general principles apply to the diagnosis and treatment of patients with suspected functional neoplasms of the endocrine pancreas. One must first recognize the abnormal physiology or characteristic syndrome. Patients are often misdiagnosed or have their symptoms disregarded for years before an accurate diagnosis is reached. Characteristic clinical syndromes are well described for insulinoma, gastrinoma, VIPoma, and glucagonoma. The somatostatinoma syndrome is nonspecific, much more difficult to recognize, and exceedingly rare. Second is the detection of hormone elevations in the serum by radioimmunoassay. Such assays are readily available for measuring insulin, gastrin, vasoactive intestinal peptide (VIP), and glucagon. Assays for somatostatin, pancreatic polypeptide (PP), prostaglandins, and other hormonal markers are less commonly available but can be obtained from certain laboratories. The third step involves localizing and staging the tumor in preparation for possible operative intervention (Algorithm 56-1).
LOCALIZATION AND STAGING
Computed Tomography
4 The initial imaging technique used to localize a PEN and stage the disease is high-quality multidetector three-dimensional CT.50 The accuracy of CT in detecting primary PENs ranges from 64% to 82% and depends largely on the size of the tumor.51,52 PENs are typically hyperdense (bright) on arterial phases of imaging. Lesions that are obvious during the early arterial phase can become isodense on later phases of imaging. Therefore, a multiphase approach is typically recommended.53,54 CT is useful in assessing size and location of the primary tumor, proximity to visceral vessels, peripancreatic lymph node involvement, and the presence or absence of liver metastases (Fig. 56-1).
Magnetic Resonance Imaging
MRI is increasingly used in the detection of PENs, particularly small lesions. They are especially well visualized on T1- and T2-weighted images with fat suppression. MRI has the advantage of increased soft tissue contrast without the administration of intravenous contrast when compared to CT.42 PENs characteristically have high signal intensity on T2-weighted images.55 On dynamic contrast-enhanced T1-weighted images, the tumors show the same typical enhancement pattern as on CT scan. The sensitivity of MRI has been reported to be between 74% and 100%.51,52
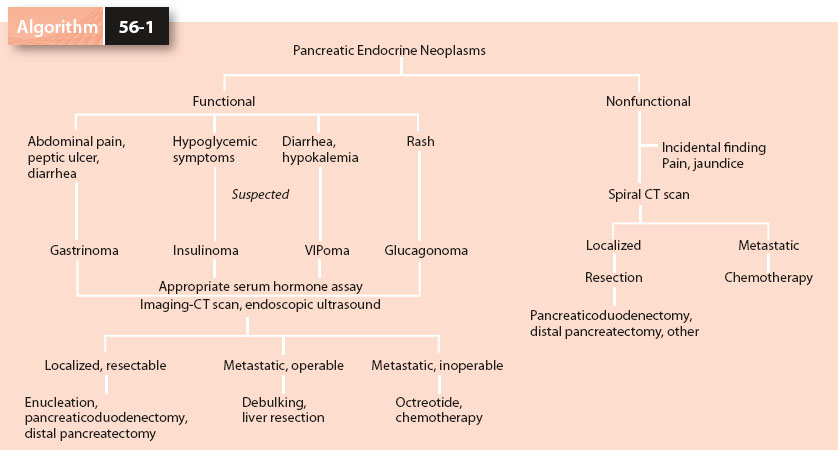
Algorithm 56-1. Diagnosis and management of pancreatic endocrine neoplasms.
Somatostatin Receptor Scintigraphy (Octreoscan)
Somatostatin receptor scintigraphy (SRS) also plays an important role in imaging patients with pancreatic endocrine tumors.56–62 In this technique, the octapeptide analog of somatostatin (Octreotide) labeled with indium-111 is administered intravenously to patients in whom a PEN is suspected. Because neuroendocrine tumors often express large numbers of somatostatin receptors on their cell surfaces (Fig. 56-2), the tracer preferentially identifies tumors. The overall sensitivity of SRS has been reported to range from 74% to near 100% depending on the functional type of PEN.63 There is a significant false-negative rate, indicating that negative SRS findings in patients with PENs should be viewed with caution. Nonfunctional tumors and insulinomas seem to be localized less frequently by SRS, while SRS performs well for gastrinoma, VIPoma, and glucagonoma. In addition, SRS appears to play a role in the evaluation of patients with metastatic pancreatic endocrine tumors, especially in identifying extrahepatic tumor spread. In a study by Frilling et al.,62 54% of patients with liver metastases had extrahepatic tumor spread detected by SRS that was not detected by alternate imaging techniques.
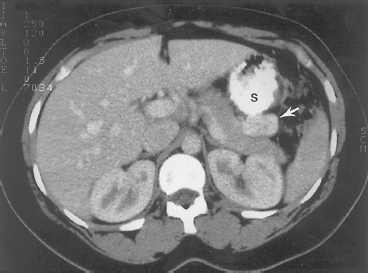
Figure 56-1. Computed tomography with oral and intravenous contrast in a patient with biochemical evidence of insulinoma. The neoplasm (arrow) is seen as a contrast-enhancing structure, 3 cm in diameter, in the tail of the pancreas posterior to the stomach (S). (From Yeo CJ. Islet cell tumors of the pancreas. In: Niederhuber JE, ed. Current Therapy in Oncology. St. Louis, MO: Mosby; 1993:272, with permission.)

Figure 56-2. Octreotide scan (anterior view) in a patient with a large endocrine tumor in the tail of the pancreas (large dark mass, upper right) and several hepatic metastases (upper left quadrant). A small amount of the tracer is seen in the bladder (lower midline).
Endoscopic Ultrasound
5 Endoscopic ultrasonography (EUS) has also shown utility in localizing PENs.64–68 Rosch et al.67 were able to localize 32 of 39 tumors (82%) correctly with EUS after CT had failed to locate the tumor (Fig. 56-3). In their experience, EUS was more sensitive than the combination of CT and visceral angiography. A more recent study by Proye et al.69 evaluated preoperative EUS and SRS in 41 patients with insulinoma and gastrinoma. The sensitivity and positive predictive value of EUS were 77% and 94%, respectively, for pancreatic tumors; 40% and 100%, respectively, for duodenal gastrinomas; and 58% and 78%, respectively, for metastatic lymph nodes. These results indicate that EUS is best at detecting lesions in the head of the pancreas. It is less successful at evaluating the distal pancreas and the duodenal wall. Additionally, the procedure is operator dependent.70 These results have been duplicated by others and have led some to suggest that EUS should serve as the initial localization procedure in patients with insulinoma and gastrinoma. Of note, the drawback to EUS is that it does not evaluate accurately for hepatic metastatic disease; rather, it is more sensitive than CT for imaging the duodenal wall, pancreatic parenchyma, and peripancreatic lymph nodes.
Intraoperative Ultrasound
Historically, the primary methods of localizing PENs intraoperatively have been visualization and palpation. With the advent of laparoscopic exploration for PENs, intraoperative ultrasound has been substituted for palpation. Results have been promising, with sensitivities reported between 75% and 90%.71,72

Figure 56-3. Endoscopic ultrasonographic image from a patient with an insulinoma (arrows) in the body of the pancreas. SV, splenic vein. (From Rosch T, Lightdale CJ, Botet JF, et al. Localization of pancreatic endocrine tumors by endoscopic ultrasonography. N Engl J Med 1992;326:1721–1726, with permission.)

Figure 56-4. Schematic depiction of data from percutaneous transhepatic portal venous sampling (PTPVS) in a patient with an insulinoma. Insulin levels are given in microunits per milliliter. These data localize the neoplasm to the head of the pancreas. (Adapted from Norton JA, Sigel B, Baker AR, et al. Localization of an occult insulinoma by intraoperative ultrasonography. Surgery 1985;97:381–384.)
Venous Sampling
Percutaneous transhepatic portal venous sampling (PTPVS) and arterial stimulation with venous sampling (ASVS) are two techniques that are used exclusively for the diagnosis and localization of PENs. In a small number of cases, CT, MRI, SRS, and EUS are unsuccessful at localizing a PEN. When insulinoma or gastrinoma are suspected, PTPVS may help in localizing the occult neoplasm.73–77 The technique involves placing a catheter percutaneously through the liver into the portal vein and then sequentially sampling for hormone levels in the splenic vein, superior mesenteric vein, and portal vein, thereby regionalizing the location of hormone production (Fig. 56-4). The overall accuracy of this test ranges from 70% to greater than 95% depending on the number of samples obtained, the persistence of autonomous hormone production by the tumor, and the careful handling and assaying of all samples. ASVS involves the selective visceral arterial injection of secretin or calcium with concurrent hepatic venous sampling for either gastrin or insulin.78,79 Gastrinoma cells are known to respond to secretin by releasing gastrin,80,81 and insulinoma cells are known to respond to calcium by releasing insulin. The provocative secretogogue is serially injected through an arterial catheter into at least three sites – the splenic, gastroduodenal, and inferior pancreaticoduodenal arteries. Samples are drawn from a hepatic vein catheter before and immediately after each injection. The arterial supply to the occult tumor can then be deduced based on which selective secretogogue injection is followed by a large increase in hepatic vein hormone concentration (Fig. 56-5). This technique, particularly when combined with intraoperative ultrasonography, results in a sensitivity of greater than 90%, essentially obviating the need for blind resection in unlocalized insulinomas.71,82 Additionally, ASVS can differentiate the 5% of patients with nesidioblastosis from those with insulinoma.83
SURGICAL EXPLORATION
At the time of surgical exploration for PEN, a complete evaluation of the pancreas and peripancreatic regions is performed. The body and tail of the pancreas are exposed by dividing the gastrocolic ligament and entering the lesser sac. This portion of the pancreas can be partially elevated out of the retroperitoneum by dividing the inferior retroperitoneal attachments to the gland. After the second portion of the duodenum has been elevated out of the retroperitoneum by means of the Kocher maneuver, the pancreatic head and uncinate process are palpated bimanually. The liver is carefully assessed for evidence of metastatic disease. Potential extrapancreatic sites of tumor are evaluated in all cases, with particular attention paid to the duodenum, splenic hilum, small intestine and its mesentery, peripancreatic lymph nodes, and reproductive tract in women. The goals of surgical therapy for PENs include controlling the symptoms of hormone excess, safely resecting maximal tumor mass, and preserving maximal pancreatic parenchyma. Management strategies, including preoperative, intraoperative, and postoperative considerations, vary for the different types of endocrine neoplasms of the pancreas.

Figure 56-5. Graphic depiction of the results of arterial stimulation with venous sampling (ASVS) in a patient with gastrinoma. The rise in hepatic vein gastrin concentration (gastrin gradient) is plotted on the y-axis, and basal values are plotted on the x-axis: 1, 100% rise; 2, 200% rise; and so forth. A rise in the hepatic vein gastrin concentration observed after the injection of secretin into the superior mesenteric artery (SMA) and gastroduodenal artery (GDA) localizes the neoplasm to the head of the pancreas or duodenum. SPL, splenic artery. (Adapted from Thom AK, Norton JA, Doppman JL, et al. Prospective study of the use of intra-arterial secretin injection and portal venous sampling to localize duodenal gastrinomas. Surgery 1992;112:1002–1028; discussion 1008–1009.)
INSULINOMA
Insulinoma is the most common functional neoplasm of the endocrine pancreas (Table 56-5). The insulinoma syndrome is associated with the following features, known as Whipple triad84:
1. Symptoms of hypoglycemia during fasting
2. Documentation of hypoglycemia, with a serum glucose level typically below 50 mg/dL
3. Relief of hypoglycemic symptoms following administration of exogenous glucose
6 Autonomous insulin secretion in insulinomas leads to spontaneous hypoglycemia, with symptoms that can be classified into two groups (Table 56-5). Neuroglycopenic symptoms include confusion, seizure, obtundation, personality change, and coma. Hypoglycemia-induced symptoms, related to a surge in catecholamine levels, include palpitations, trembling, diaphoresis, and tachycardia. In most cases, patients consume carbohydrate-rich meals and snacks to relieve or prevent these symptoms.
Table 56-5 Insulinoma

Whipple triad is not specific for insulinoma. The differential diagnosis of adult hypoglycemia is extensive and includes the following: reactive hypoglycemia, functional hypoglycemia associated with gastrectomy or gastroenterostomy, nonpancreatic tumors, pleural mesothelioma, sarcoma, adrenal carcinoma, hepatocellular carcinoma, carcinoid, hypopituitarism, chronic adrenal insufficiency, extensive hepatic insufficiency, and surreptitious self-administration of insulin or ingestion of oral hypoglycemic agents.
A common error made in evaluating a patient with suspected insulinoma is to begin with an oral glucose tolerance test. Instead, insulinoma is most reliably diagnosed by means of a monitored fast (via the withholding of exogenous glucose). During a monitored fast, blood is sampled for glucose and insulin determinations every 4 to 6 hours and when symptoms appear. Hypoglycemic symptoms typically occur when glucose levels are below 50 mg/dL, with concurrent serum insulin levels often exceeding 25 microunits/mL. Additional support for the diagnosis of insulinoma comes from the calculation of the insulin-to-glucose ratio at different times during the monitored fast. Normal persons have insulin-to-glucose ratios below 0.3, whereas patients with insulinoma typically demonstrate insulin-to-glucose ratios above 0.4 after a prolonged fast. Other measurable β-cell products synthesized in excess in patients with insulinoma include C peptide and proinsulin. Elevated levels of both are typically found in the peripheral blood of patients with insulinoma.
The possibility of the surreptitious administration of insulin or oral hypoglycemic agents should be considered in all patients with suspected insulinoma. Levels of C peptide and proinsulin are not elevated in patients who self-administer insulin. Additionally, patients self-administering either bovine or porcine insulin may demonstrate anti-insulin antibodies in circulating blood. The ingestion of oral hypoglycemic agents, such as sulfonylureas, can be assessed by means of standard toxicologic screening.
7 Insulinomas are evenly distributed throughout the pancreas, with one-third found in the head and uncinate process, one-third in the body, and one-third in the tail of the gland.85 Less than 3% are located outside the pancreas, with these lesions located in the peripancreatic area.86 Ninety percent are found to be benign solitary adenomas amenable to surgical cure. Ninety percent of insulinomas are sporadic, with approximately 10% being associated with the MEN-1 syndrome. In patients with MEN-1, the possibility of multiple insulinomas must be considered, and recurrence rates are higher. In approximately 10% of patients, insulinoma is metastatic to the peripancreatic lymph nodes or liver, making the diagnosis of malignant insulinoma.

Figure 56-6. The technique for enucleating a benign pancreatic endocrine neoplasm with scissors (A) or electrocautery (B). C: After enucleation, the site of excision is drained.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


