Neoplasia
Jacquelyn L. Banasik
Key Questions
• How do neoplastic cells differ from normal cells?
• In what ways do benign and malignant tumors differ?
• How might overexpression of proto-oncogenes lead to abnormal cellular proliferation?
• How might underexpression of tumor suppressor genes lead to abnormal cellular proliferation?
• How are tumor grading and staging used to guide the selection of cancer therapies?
• How might lifestyle and carcinogen exposure contribute to cancer risk?
• What treatment options are available for benign and malignant tumors?

http://evolve.elsevier.com/Copstead/
Neoplasia means “new growth.” In common use, the term implies an abnormality of cellular growth and may be used interchangeably with the term tumor. It is no surprise that the discovery of a tumor in an individual can evoke feelings of disbelief, anger, and dread. Characterization of the tumor cells is of critical importance to determine whether the tumor is benign or malignant. The term cancer is applied only to malignant neoplasms. The diagnosis of a benign growth is received with great relief inasmuch as the tumor is generally easily cured. The diagnosis of a malignant cancer, on the other hand, may herald months of intensive and often uncomfortable treatment with uncertain outcomes. Cancer remains the second leading cause of death in the United States for both men and women.
It is increasingly clear that cancer is associated with altered expression of cellular genes that normally regulate cell proliferation and differentiation. A unified theory of cancer causation has emerged, and new methods for cancer therapy continue to be developed. Cancer is a complex, multifaceted disorder with each individual cancer having some unique properties. A better understanding of the molecular characteristics of individual cancers is encouraging the development of specific therapies that target each cancer’s weaknesses.
Benign Versus Malignant Growth
Characteristics of Benign and Malignant Tumors
The terms benign and malignant refer to the overall consequences of a tumor to the host. Generally, malignant tumors have the potential to kill the host if left untreated, whereas benign tumors do not. This difference is not strict because some benign tumors may be located in critical areas. For example, a benign tumor may be life threatening if it causes pressure on the brain or blocks an airway or blood vessel. Histologic examination of a tumor is the primary mode for determining its benign or malignant nature. Certain tumor characteristics have historically been shown to indicate malignant potential. Important considerations include localization of the tumor and determination of the degree of tumor cell differentiation.
Benign tumors do not invade adjacent tissue or spread to distant sites. Many benign tumors are encapsulated by connective tissue, which is an indication of strictly local growth. Any evidence that tumor cells have penetrated local tissues (invasiveness), lymphatics, or blood vessels suggests a malignant nature with potential to spread to distant sites (metastasize).
As a general rule, benign cells more closely resemble their tissue type of origin (e.g., skin, liver) than do malignant cells. The degree of tissue-specific differentiation has traditionally been used to predict malignant potential. A lack of differentiated features in a cancer cell is called anaplasia, and a greater degree of anaplasia is correlated with a more aggressively malignant tumor.1 Anaplasia is indicated by variation in cell size and shape within the tumor, enlarged nuclei, abnormal mitoses, and bizarre-looking giant cells (Figure 7-1). Regardless of histologic appearance, invasion of local tissue or evidence of metastasis to distant sites confirms the diagnosis of malignancy.
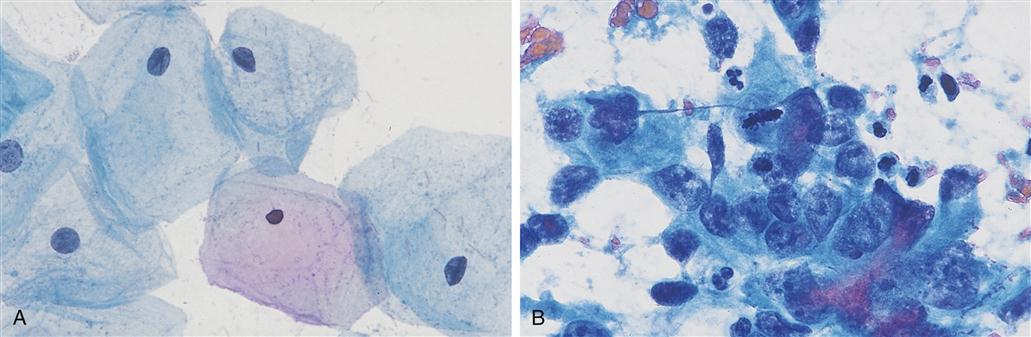
Other differences between benign and malignant tumors have been noted (Table 7-1). Benign tumors generally grow more slowly, have little vascularity, rarely have necrotic areas, and often retain functions similar to those of the tissue of origin. Conversely, malignant tumors often grow rapidly and may initiate vessel growth in the tumor. They frequently have necrotic areas and are dysfunctional.
TABLE 7-1
GENERAL CHARACTERISTICS OF BENIGN AND MALIGNANT TUMORS
| CHARACTERISTIC | BENIGN | MALIGNANT |
| Histology | Typical of tissue of origin Few mitoses | Anaplastic, with abnormal cell size and shape Many mitoses |
| Growth rate | Slow | Rapid |
| Localization/metastasis | Strictly local, often encapsulated/no metastasis | Infiltrative/frequent metastases |
| Tumor necrosis | Rare | Common |
| Recurrence after treatment | Rare | Common |
| Prognosis | Good, unless in critical area | Poor if untreated |
Tumor Terminology
General rules for the naming of tumors have been developed to indicate the tissue of origin and the benign or malignant nature of the tumor. The suffix -oma is used to indicate a benign tumor, whereas carcinoma and sarcoma are used to indicate malignant tumors. Carcinoma refers to malignant tumors of epithelial origin and sarcoma to malignant tumors of mesenchymal (nerve, bone, muscle) origin. Thus a benign tumor of glandular tissue would be called an adenoma, but a malignant tumor of the same tissue would be called an adenocarcinoma (Table 7-2). Some notable exceptions to the rules are lymphomas, hepatomas, and melanomas, which are all highly malignant despite their -oma suffix. Leukemia refers to a malignant growth of white blood cells. The great majority of human cancers (90%) are carcinomas from malignant transformation of epithelial cells.2
TABLE 7-2
NOMENCLATURE FOR NEOPLASTIC DISEASES
From Murphy GP, Lawrence W, Lenhard RE, editors: American Cancer Society textbook of clinical oncology, Atlanta, 1995, Author, p 77. Reproduced by permission of the American Cancer Society.
The Malignant Phenotype
Cells growing in normal tissue have predictable relationships with neighboring cells. In a particular tissue, the rate of cell proliferation is precisely matched to the rate of cell death. Normal cells require constant reassurance in the form of survival signals from their environment that their continued existence is desirable, and they proliferate only when space is available and appropriate mitogen-stimulating signals are present. Normal cells also respond to signals instructing them to actively destroy themselves in a process called apoptosis (see Chapter 4). Cancer cells, however, do not obey the rules; they have escaped the normal mechanisms of growth control. A number of antisocial properties develop in malignant cells that allow them to proliferate at the expense of other cells and tissues of the body. These abnormal behaviors can be summarized as follows:
• Cancer cells proliferate despite lack of growth-initiating signals from the environment.
• Cancer cells invade their local tissue and overrun their neighbors.
Most cancers are thought to arise from stem cells that are present in tissues. Tissue stem cells are capable of unlimited proliferation, entering the cell cycle to produce two daughter cells—with one cell retaining the original stem cell properties and the other becoming a more differentiated cell, but still capable of proliferation. Normally, the partially differentiated cells can undergo only a limited number of cell divisions before they permanently leave the cell cycle and become senescent.2 Either a stem cell or a partially differentiated cell has the potential to acquire the genetic mutations necessary to become malignant.
Epidemiology and Cancer Risk Factors
Cancer accounts for approximately 25% of all deaths, which makes it the second leading cause of death in the United States. Most cancer deaths (77%) occur in persons older than 55 years. The American Cancer Society (ACS) estimates that men have almost a 1 in 2 lifetime risk of developing cancer and women have slightly higher than a 1 in 3 risk. The 5-year relative survival rate for all cancers combined is about 68%.3 The 5-year survival rate does not distinguish between those who were cured and those who have relapsed or are still in treatment. Fortunately, the current view of cancer causation predicts that many cancers are preventable. Indeed, one third of cancer- related deaths may be attributed to lifestyle factors. Lifestyle factors of particular importance are tobacco use, nutrition, and obesity.4 Sun exposure is a significant risk factor for skin cancer (Chapter 53), and sexual exposure to certain strains of human papillomavirus predisposes to cervical cancer (Chapter 34). The high incidence and relative ease of screening for breast, cervical, colorectal, and prostate cancers has prompted the development of guidelines for early detection of these cancers. The current recommendations for early detection of cancer in average-risk, asymptomatic persons are shown in Table 7-3. Statistics regarding some of the major forms of cancer are shown in Figure 7-2. Further discussions of particular cancers can be found in chapters relating to corresponding body systems.
TABLE 7-3
SCREENING GUIDELINES FOR THE EARLY DETECTION OF CANCER IN AVERAGE-RISK ASYMPTOMATIC PEOPLE
| CANCER SITE | POPULATION | TEST OR PROCEDURE | FREQUENCY |
| Breast | Women, age 20+ | Breast self-examination | Beginning in their early 20s, women should be told about the benefits and limitations of breast self-examination (BSE). The importance of prompt reporting of any new breast symptoms to a health professional should be emphasized. Women who choose to do BSE should receive instruction and have their technique reviewed on the occasion of a periodic health examination. It is acceptable for women to choose not to do BSE or to do BSE irregularly. |
| Clinical breast examination | For women in their 20s and 30s, it is recommended that clinical breast examination (CBE) be part of a periodic health examination, preferably at least every 3 years. Asymptomatic women aged 40 and over should continue to receive a clinical breast examination as part of a periodic health examination, preferably annually. | ||
| Mammography | Begin annual mammography at age 40–50∗ | ||
| Colorectal† | Men and women, age 50+ | Tests that find polyps and cancer: Flexible sigmoidoscopy‡ or | Every 5 years starting at age 50 |
| Colonscopy, or | Every 10 years, starting at age 50 | ||
| Double-contrast barium enema (DCBD) ‡ | Every 5 years, starting at age 50 | ||
| Tests that mainly find cancer: | Annual, starting at age 50 | ||
| Stool DNA test (sDNA)‡ | Interval uncertain, starting at age 50 | ||
| Prostate | Men, age 50+ | Prostate-specific antigen test (PSA) with or without digital rectal exam (DRE) | Asymptomatic men who have at least 10-year life expectancy should have an opportunity to make an informed decision with their health care provider about screening for prostate cancer after receiving information about the uncertainties, risks, and potential benefits associated with screening. Prostate cancer screening should not occur without an informed decision-making process. |
| Cervix | Women, age 21+ | Pap test | Cervical cancer screening should begin approximately 3 years after a woman begins having vaginal intercourse, but no earlier than 21 years of age. Screening should be done every year with conventional Pap tests or every 2 years using liquid-based Pap tests. At or after age 30, women who have had three normal test results in a row may get screened every 2 to 3 years with cervical cytology (either conventional or liquid-based Pap test) alone, or every 3 years with an HPV DNA test plus cervical cytology. Women 70 years of age and older who have had three or more normal Pap tests and no abnormal Pap tests in the past 10 years and women who have had a total hysterectomy may choose to stop cervical cancer screening. |
| Endometrial | Women, at menopause | At the time of menopause, women at average risk should be informed about risks and symptoms of endometrial cancer and strongly encouraged to report any unexpected bleeding or spotting to their physicians. | |
| Cancer-related checkup | Men and women age 20+ | On the occasion of a periodic health examination, the cancer-related checkup should include examination for cancers of the thyroid, testicles, ovaries, lymph nodes, oral cavity, and skin, as well as health counseling about tobacco, sun exposure, diet and nutrition, risk factors, sexual practices, and environmental and occupational exposures. | |
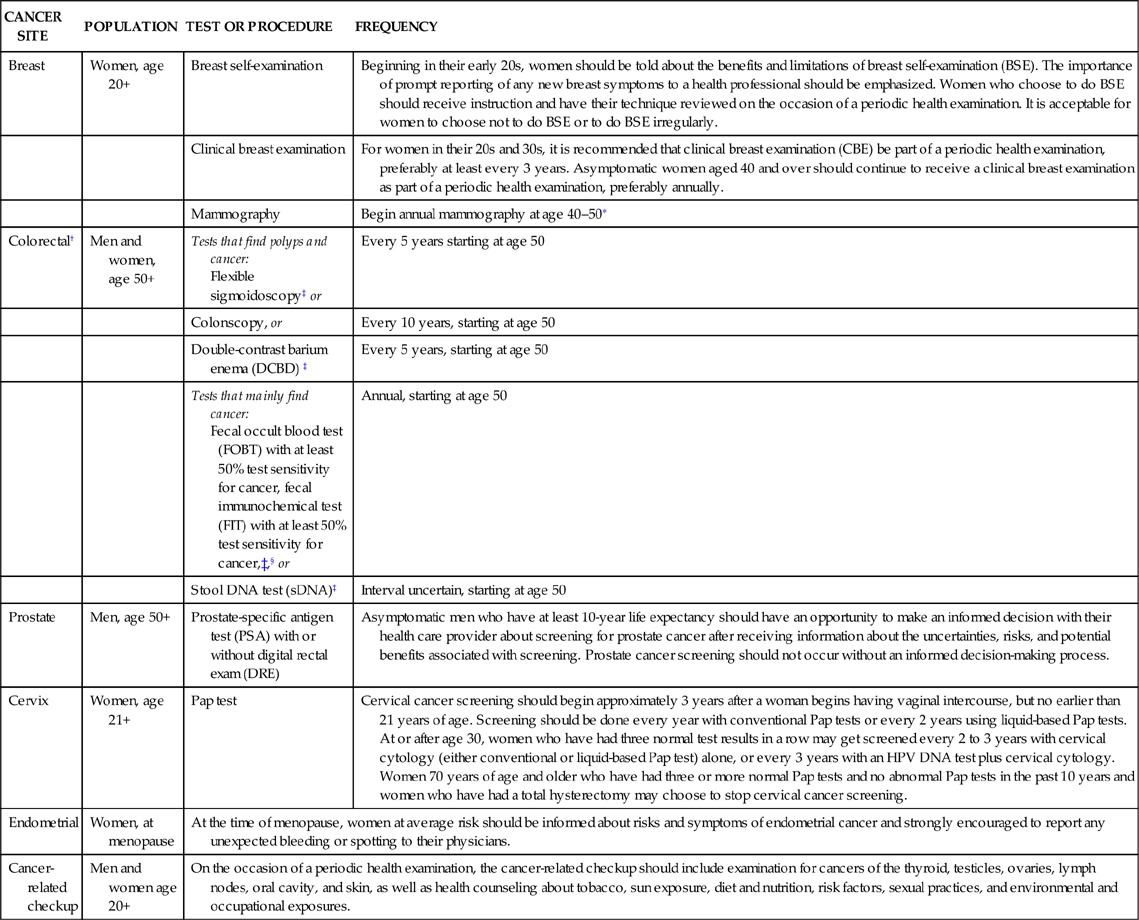
∗Beginning at age 40, annual clinical breast examination should be performed before mammography.
†Individuals with a personal or family history of colorectal cancer or adenomas, inflammatory bowel disease, or high-risk genetic syndromes should continue to follow the most recent recommendations for individuals at increased or high risk.
‡Colonoscopy should be done if test results are positive.
§For FOBT or FIT used as a screening test, the take-home multiple sample method should be used. An FOBT or FIT done during a digital rectal exam in the doctor’s office is not adequate for screening.
American Cancer Society: Cancer facts and figures—2012, Atlanta, 2012, American Cancer Society; ACOG Committee on Practice Bulletins-Gynecology. Obstet Gynecol 114(6):1409-1429, 2009.
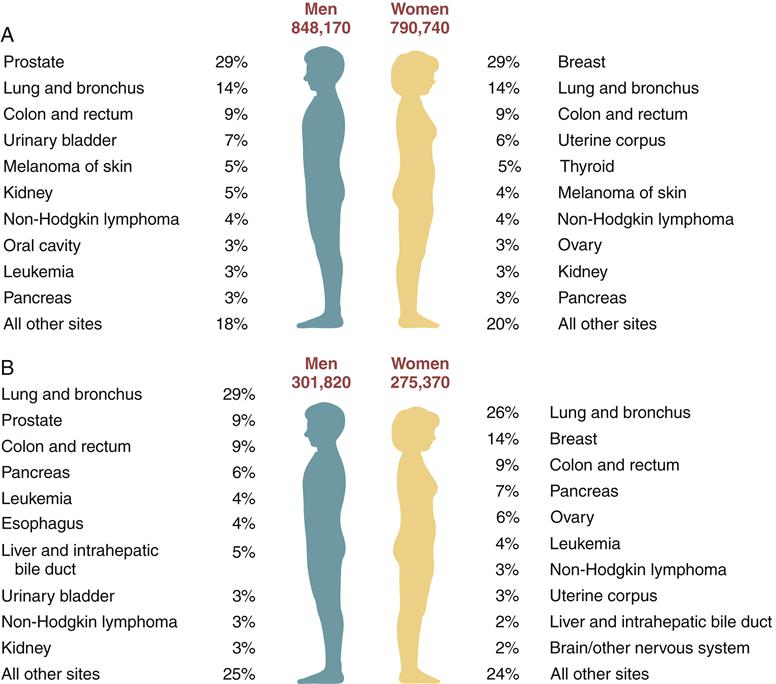
Excludes basal and squamous cell skin cancers and in situ carcinomas except urinary bladder. (American Cancer Society: Cancer facts and figures—2012, Atlanta, 2012, American Cancer Society.)
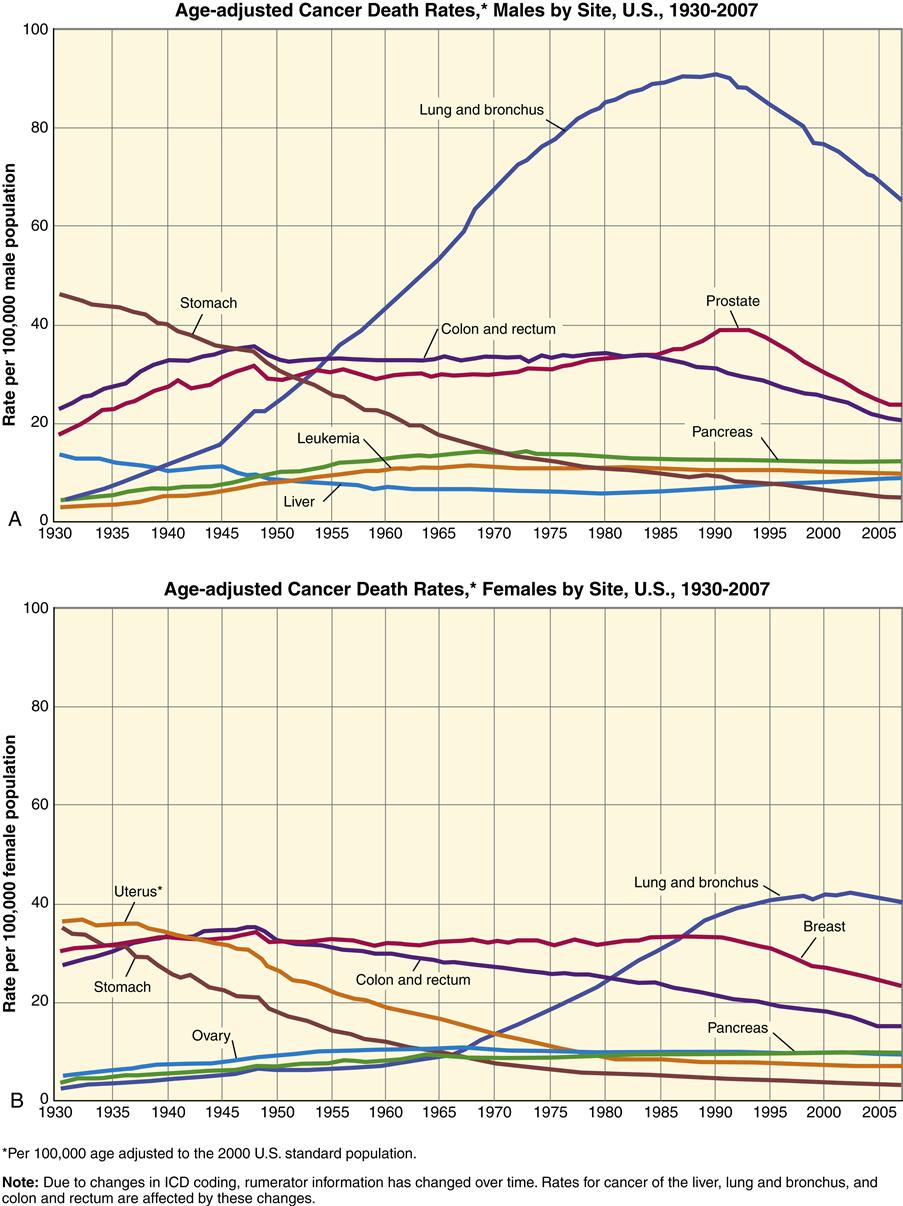
Tobacco Use
The impact of tobacco use on cancer-related death can be most vividly seen by looking at cancer death rates in the United States from 1930 to 2007 (Figure 7-3). Whereas all other cancer-related death rates declined or remained relatively stable, the death rate from lung cancer increased dramatically. The increase is attributable almost entirely to smoking. Lung cancer remains the leading cause of cancer death in both men and women, accounting for 30% of all cancer deaths. Lung cancer has one of the worst survival rates of all cancers—only 15%. In addition to lung cancer, tobacco use has been linked with cancer of the pancreas, bladder, kidney, mouth, esophagus, and cervix (Figure 7-4). Smoking prevalence among adults in the United States declined from 42% in 1965 to 21% in 2004 and has remained stable at 21%, with rates being approximately 5% lower in women than in men. An estimated 45 million U.S. adults currently smoke cigarettes. Approximately 20% of high school students reported being cigarette smokers in 2009. In 2006, only 8% of college graduates were current smokers, compared to 21% in 1983.3
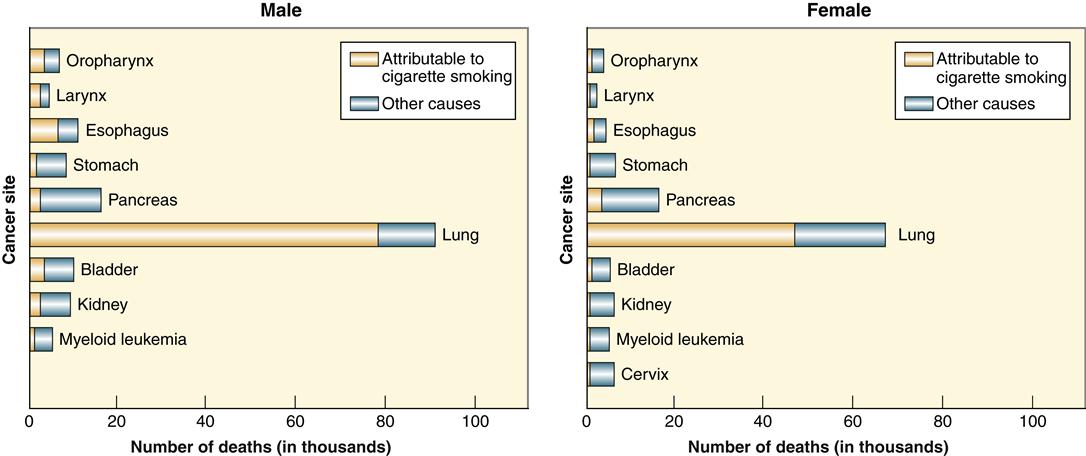
Carcinogens can be grouped into two major types: those that cause genetic damage (initiators) and those that promote growth of the tumor (promoters). Tobacco smoke contains hundreds of compounds, many of which have known genotoxicity (e.g., polycyclic aromatic hydrocarbons, nicotine derivatives) and probably serve as initiators. Tobacco smoke also contains promoters, which spur the mutant cells to proliferate. Second-hand smoke contains more than 7000 chemicals, of which 69 are known to cause cancer.3 The American Cancer Society estimates that about 3400 nonsmoking adults die from lung cancer each year as a result of exposure to second-hand smoke.3
Nutrition
The scientific study of nutrition and cancer is complex, and it is not clear how single nutrients, combinations of nutrients, overnutrition and energy imbalance, or the amount and distribution of body fat affect a person’s risk for specific cancers.3 The ACS suggests a mostly plant-based diet emphasizing a variety of vegetables, fruits, and whole grains. The ACS endorses limiting the intake of red and processed meats, while controlling total caloric intake to maintain a healthy weight. Individual nutritional supplements are not recommended for cancer prevention.3 The results of randomized clinical trials of antioxidant supplements and selenium have shown no reduction in risk for cancer, at least in generally well-nourished populations.3
Fat
Several epidemiologic studies performed in the 1970s and early 1980s suggested a relationship between high-fat diets and the development of breast, colon, and prostate cancer. In some studies, however, higher fat intake was found to be protective against some cancers. A pooled analysis of seven large studies found no link between fat intake and the risk of breast cancer.5 The results of one large, randomized clinical trial investigating the effect of a low-fat diet on the occurrence of invasive breast cancer found no difference between the control and low-fat groups after 8 years of follow-up in postmenopausal women.6 Fat or calorie intake and high production of insulin may affect breast cancer outcomes depending on tumor cell type and hormone responsiveness. Further research on the specific type of fat intake and other cofactors is needed to clarify the fat-cancer relationship. Several studies in animals have shown that regardless of fat intake, tumor growth may be inhibited by caloric restriction. Some investigators have proposed a link between high insulin production and breast cancer.
Fiber
Fiber is a general term for nondigestible dietary substances that remain in the intestinal lumen, increase fecal bulk, and improve bowel regularity. Fiber includes a diversity of compounds such as cellulose, bran, and pectin. An association between fiber intake and colorectal cancer proposed in the early 1970s was based on a study comparing the incidence of certain ailments in Americans and Africans.7 A number of correlational and comparison studies done since that time have yielded conflicting results, and large randomized trials failed to show a benefit.8 Part of the difficulty may be linked to the way that different studies define dietary fiber. Because fiber is associated with beneficial effects on digestion and elimination, fiber intake in the range of 10 to 13 g per 1000 calories consumed is generally recommended.
Alcohol
Alcohol intake has been linked to a number of cancers, including breast, esophageal, laryngeal, and liver cancer. Alcohol may exert its cancer-promoting effects through impairment of the liver’s ability to metabolize harmful substances and endogenous hormones. Moderate alcohol intake has been shown to increase estrogen levels, which may account for its promoting effects on breast cancer.9 As a carbohydrate-dense substance, alcohol may contribute to cancer risk through its effects on insulin secretion. Insulin is a general growth factor for a number of tissues. Limiting alcohol intake may provide a modest reduction in cancer risk.
Antioxidants
Until recently, the emphasis of cancer prevention has been on the identification and avoidance of cancer-causing agents. However, increasing interest has been shown in finding substances with cancer-protective properties. The fact that DNA damage is an important step in cancer initiation, coupled with the knowledge that oxygen free radicals can impart this damage, led to the idea that antioxidants may have protective effects for cancer. The specific agents tested in clinical trials included β-carotene, vitamin E, vitamin C, selenium, retinol, zinc, riboflavin, and molybdenum. None of the completed trials produced convincing evidence to justify the use of traditional antioxidant-related vitamins or minerals for cancer prevention.10
Vitamin A and the antioxidant trio of vitamin E, β-carotene, and vitamin C have been most widely studied. The use of antioxidants to prevent cancer sounds like a good idea; however, several large-scale studies have failed to reveal a benefit and some have found that the risk of cancer may be increased.11,12 At present, it may be prudent to consume a diet high in natural fruit and vegetable sources of antioxidants.
Genetic Mechanisms of Cancer
Despite much progress in our understanding of how mechanisms of growth control and cellular differentiation may go awry, there is still no simple answer to the question, “What causes cancer?” It is increasingly evident, however, that cancer is primarily a disorder of gene expression. Early support for a genetic basis of cancer came from the observation that cancer often resulted from agents known to damage deoxyribonucleic acid (DNA). In the 1970s a number of potential cancer-causing agents (carcinogens) were identified by demonstrating their mutagenic potential.13 The suggestion that mutant genes were the basis for cancer launched intense research to identify the cancer-causing gene or genes.
Cancer-critical genes are grouped into two broad classes, according to whether overactivity of the gene contributes to cancer (gain-of-function mutations) or whether underactivity is the problem (loss-of-function mutations). Both categories of genes result in similar effects in enhancing cell proliferation and survival.2 Genes in the first category are called proto-oncogenes, which normally code for components of the cellular growth–activating pathways. A proto-oncogene in its mutant, overactive, or overexpressed form is called an oncogene. Genes in the second category of cancer-related genes are called tumor suppressor genes, which normally inhibit cell proliferation. Cancers may arise when tumor suppressor gene function is lost or abnormally inhibited. To achieve malignant transformation, a cell must generally suffer mutations in a combination of these growth regulatory genes. A cell thus transformed passes on these mutations to its progeny when it divides and forms a clone of abnormally proliferating cells. Numerous studies have begun to unravel the details of how proto-oncogenes and tumor suppressor genes may dysfunction and contribute to the malignant phenotype. In addition to the genes that regulate the cell cycle, two other categories of genes that monitor and maintain the genome contribute indirectly to the development of cancer. These are the DNA-repair genes and the genes that regulate apoptosis (see Chapter 4).
Proto-Oncogenes
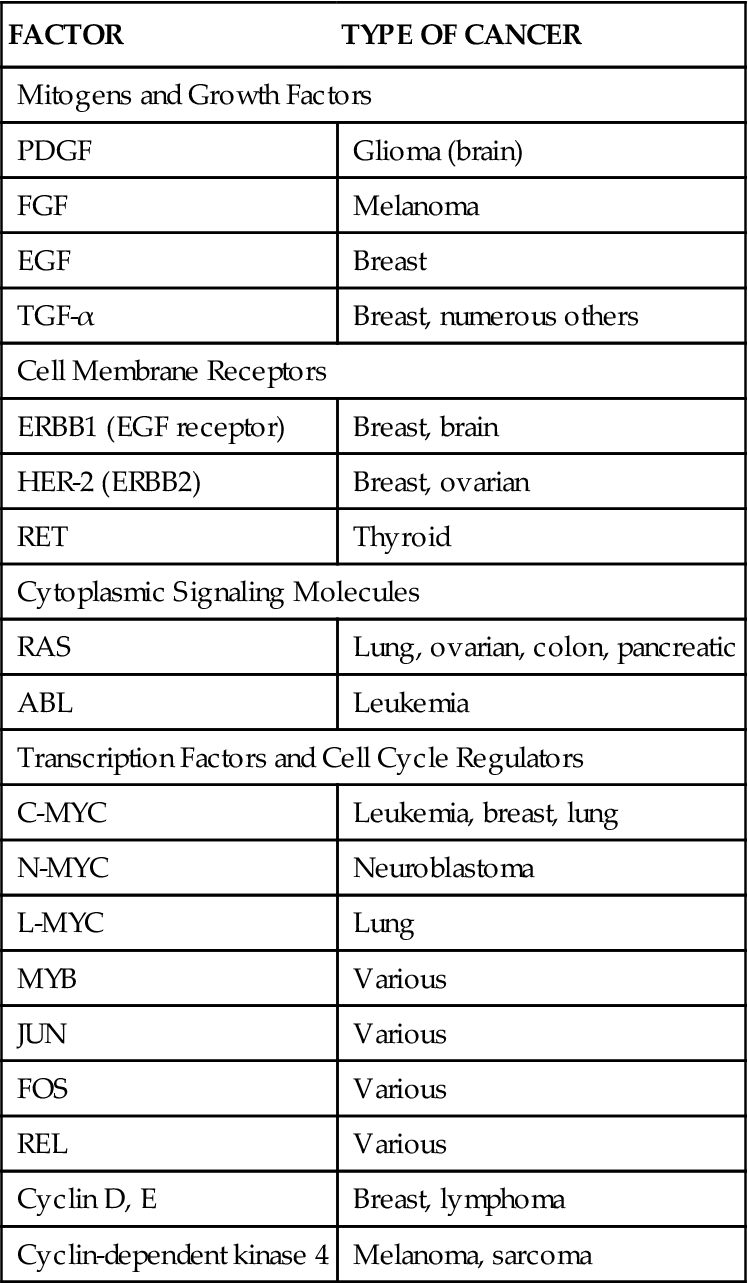
Proto-oncogenes were the first of the tumor-associated genes to be discovered, and hundreds have been described to date.2 As often happens in the study of genes, a gene associated with a disease process is identified long before its normal cellular function is elucidated. Thus genes associated with cancer are traditionally named for the cancer in which they were first discovered (in mutant form) rather than for their normal cellular function. Many of the first cancer-associated genes, called oncogenes, were initially identified in viruses and still retain the name reflecting their viral discovery. The term proto-oncogene was created to label the normal cellular gene that can be transformed into an oncogene by activating (gain-of-function) mutations. A representative list of known proto-oncogenes is shown in Table 7-4.
TABLE 7-4
EXAMPLES OF GAIN-OF-FUNCTION PROTO-ONCOGENES AND THEIR MECHANISMS OF ACTION
| FACTOR | TYPE OF CANCER |
| Mitogens and Growth Factors | |
| PDGF | Glioma (brain) |
| FGF | Melanoma |
| EGF | Breast |
| TGF-α | Breast, numerous others |
| Cell Membrane Receptors | |
| ERBB1 (EGF receptor) | Breast, brain |
| HER-2 (ERBB2) | Breast, ovarian |
| RET | Thyroid |
| Cytoplasmic Signaling Molecules | |
| RAS | Lung, ovarian, colon, pancreatic |
| ABL | Leukemia |
| Transcription Factors and Cell Cycle Regulators | |
| C-MYC | Leukemia, breast, lung |
| N-MYC | Neuroblastoma |
| L-MYC | Lung |
| MYB | Various |
| JUN | Various |
| FOS | Various |
| REL | Various |
| Cyclin D, E | Breast, lymphoma |
| Cyclin-dependent kinase 4 | Melanoma, sarcoma |
The majority of proto-oncogenes described to date code for components of cell-signaling systems that promote cell proliferation.2 These components can be grouped into four broad categories: (1) growth factors, (2) receptors, (3) cytoplasmic signaling molecules, and (4) nuclear transcription factors (Figure 7-5). Excessive activity in any of these components may release the cell from environmental feedback and allow it to proliferate abnormally.
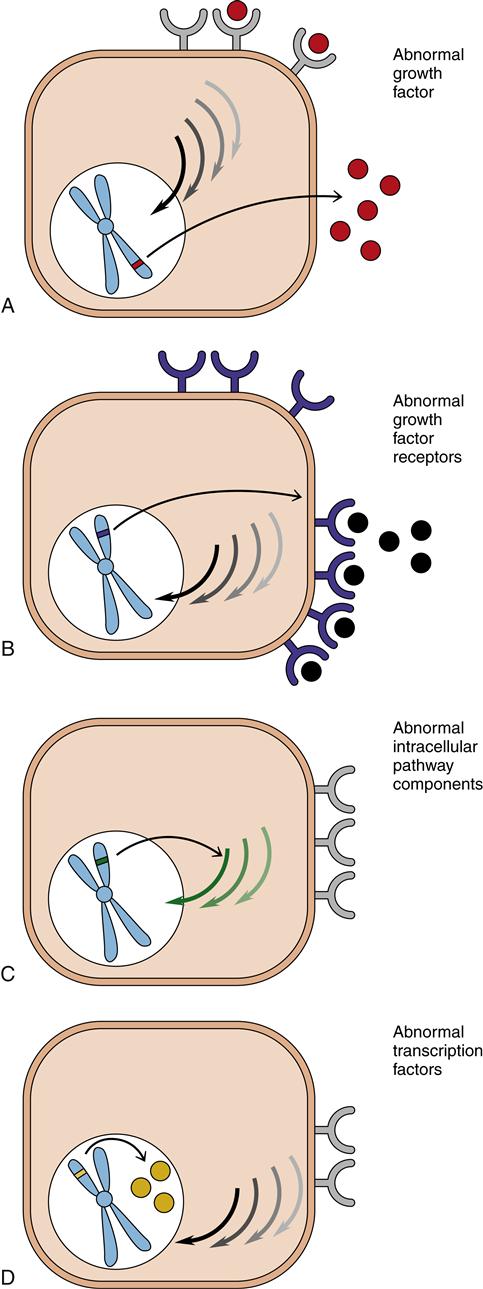
A, Production of growth factors (mitogens). B, Production of growth factor receptors. C, Intracellular pathway disturbances. D, Activation of transcription factors for growth.
Growth Factors (Mitogens)
The first proto-oncogenes to be discovered coded for growth factors. A great deal of intercellular communication is accomplished through the cell-to-cell transmission of growth factors. Growth factors are small peptides that are manufactured by cells and secreted into the extracellular space. They diffuse to nearby cells and interact with receptors on the target cell surface. Binding of growth factors to cell surface receptors activates signaling cascades within the cell that enhance proliferation. As a general principle, cells do not independently produce growth factors sufficient to stimulate their own proliferation. The proliferation signals must be produced by the cell’s environment. The cell’s environment also conveys growth-inhibiting signals. Overproduction of stimulatory growth factors by a mutant proto-oncogene can shift the balance of signals and produce excessive self-stimulated growth (autocrine signaling). Examples of tumor-secreted growth factors include platelet-derived growth factor (PDGF), transforming growth factor-α (TGF-α), and epidermal growth factor (EGF). Certain cancer types typically secrete particular growth factors. For example, platelet-derived growth factor is commonly oversecreted in glial cell cancers (brain tumors) and connective tissue cancers (sarcomas).
Growth Factor Receptors
Peptide growth factors (mitogens) cannot penetrate the cell membrane directly, so their presence at the cell surface must be transmitted intracellularly by cell surface receptors. Receptors are transmembrane proteins with the mitogen-binding area on the outside of the cell and an enzyme-activating area on the inside of the cell. These receptors are extremely specific; they will bind with only one particular mitogen. Binding activates a series of reactions within the cell that eventually leads to cell proliferation.
A mutational event may allow the expression of receptors that should not be present at all or allow excessive amounts of normally present receptors, or it may produce receptors with abnormally high affinity. All of these changes result in excessive responsiveness to the mitogens normally present in the cell’s environment. Some mutant receptors may even be active in the absence of growth factors and spur the cell to divide despite the absence of environmental signals to do so. An important example of a receptor abnormality is the overexpression of human epidermal growth factor receptor type 2 (HER2) receptors in about 25% of breast cancers. The overactive receptors stimulate proliferation of tumor cells even when there is little or no epidermal growth factor bound to them.
Cytoplasmic Signaling Pathways
A third way in which oncogenes may facilitate proliferation is by the manufacture of excessive or abnormal components of the intracellular signaling pathways. These pathways involve numerous enzymes and chemicals that normally function to transmit signals from activated receptors at the cell surface to the cell nucleus. A mutant proto-oncogene that codes for excessive or abnormal cytoplasmic signaling components could cause activation of the pathway even though no signal was received at the cell surface. The best understood example of this mechanism is mutations of the ras gene family. Proteins encoded by ras genes are monomeric G-proteins that transmit signals from receptors at the cell surface into the interior of the cell. The ras protein is active when it has guanosine triphosphate (GTP) bound to it, but it quickly hydrolyzes the GTP, thus automatically turning itself off after a brief period of activity. A mutation in the ras gene can code for a protein that is unable to hydrolyze GTP, so it remains persistently active and stimulates cell proliferation inappropriately. Mutations of the ras genes occur in about 20% of all human cancers, including leukemias and lung, ovarian, colon, and pancreatic cancer.1
Transcription Factors
The entire proliferation pathway, including the growth factor (mitogen), the receptor, and the intracellular cascade, ultimately affects transcription of a set of genes in the nucleus that spur the cell to enter the S phase. A number of proto-oncogenes have been identified that code for transcription factors in the nucleus. Transcription factors are proteins that must be assembled at the promoter area to begin gene transcription (see Chapters 3 and 5). Transcription factors are normally sequestered and prevented from indiscriminate activity until appropriate signals cause their release. Mutations in transcription factor genes may cause overproduction of transcription factors or interfere with the normal mechanisms for keeping them in check. Myc, jun, and fos are examples of proto-oncogenes that code for nuclear transcription factors. Abnormalities of the myc genes are found in numerous cancers, including lung and breast cancer, leukemia, and neuroblastoma.
From Proto-Oncogene to Oncogene
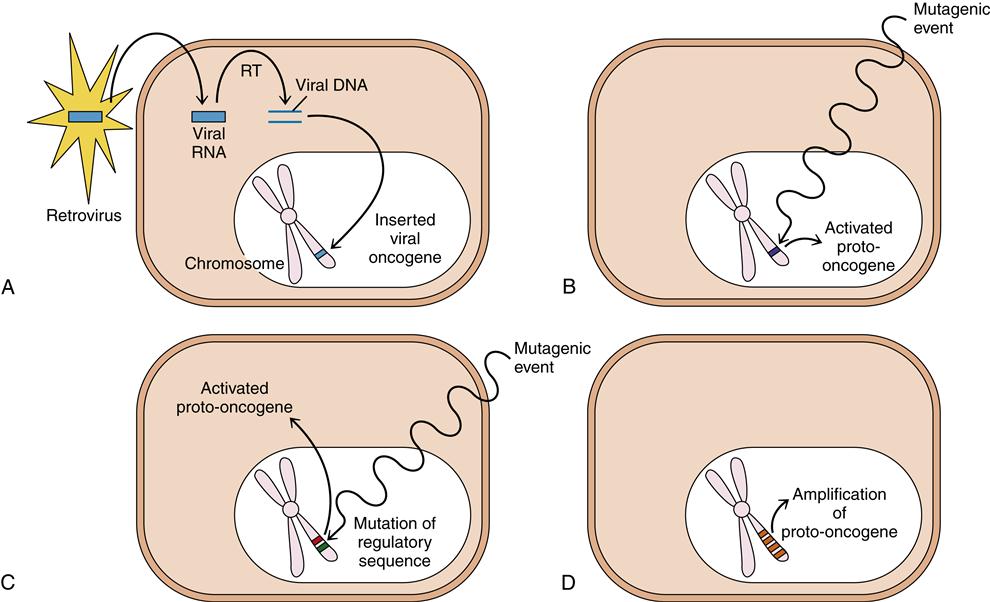
Proto-oncogenes become activated oncogenes when mutations alter their activity so that proliferation-promoting signals are generated inappropriately. At least four general ways in which proto-oncogenes can be activated are known (Figure 7-6): (1) Oncogenes may be introduced into the host cell by a retrovirus; (2) a proto-oncogene within the cell may suffer a mutagenic event that changes its structure and function; (3) a DNA sequence that normally regulates proto-oncogene expression may be damaged or lost and allow the proto-oncogene to become abnormally active; and (4) an error in chromosome replication may cause extra copies of the proto-oncogene to be included in the genome (amplification).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


