INTRODUCTION
LEARNING OBJECTIVES
After studying this chapter you should understand:
The pathogenic role of specific tyrosine kinase mutations in various myeloproliferative disorders.
The role of BCR-ABL in the diagnosis and targeted therapy of chronic myelogenous leukemia.
The diagnostic and pathogenic role of JAK2 mutations in polycythemia vera.
The pathophysiology and clinical features of the myelodysplastic syndromes.
CANCER AS A GENETIC DISEASE
Like other cancers, hematologic malignancies are caused by acquired somatic mutations that activate proto-oncogenes or inactivate tumor suppressors. Remarkably, we now know that at least some of these potentially oncogenic events occur from time to time in the hematologic cells of most if not all healthy people, the vast majority of whom will not suffer from a hematologic malignancy during their lifetimes. It follows that no single mutation is sufficient to create these cancers. Instead, they appear to result from the interplay of multiple mutations that collaborate to create a transformed phenotype. Molecular analyses have proven that although tumor cells within an individual cancer demonstrate some degree of genetic heterogeneity, all share a core set of identical mutations, indicating that they originate from a single, transformed cell.
The appearance and behavior of specific hematologic malignancies stem from two major factors: 1) the identity of the oncogenic mutations found in the tumor cells and 2) the cellular context, which is to say the identity of the founding cell from which the tumor originates. The latter dictates the effects of the former on cellular proliferation, survival, and differentiation. These two themes—acquired mutations and cellular context—will be woven through our discussion of all of the hematologic malignancies. In this chapter, we discuss the myeloproliferative disorders and myelodysplastic syndromes, tumors that originate in early hematopoietic progenitors that primarily affect marrow function and give rise to differentiated progeny. Subsequent chapters will cover the acute leukemias, aggressive tumors of early myeloid or lymphoid progenitors, and the diverse collection of tumors that are derived from mature lymphocytes and plasma cells.
MYELOPROLIFERATIVE DISORDERS
The molecular hallmark of the myeloproliferative disorders is the presence of gain-of-function mutations that activate tyrosine kinases, enzymes that phosphorylate proteins on tyrosine residues. In general, these aberrant tyrosine kinases turn on the same pathways that are normally activated by hematopoietic growth factors, but they do so in a growth factor–independent fashion. In Chapter 2, we noted that the pathways activated by hematopoietic growth factors enhance the growth and survival of myeloid progenitors in the bone marrow. As a result of constitutive activation of pro-growth and pro-survival signaling pathways, the bone marrow in the myeloproliferative disorders is hypercellular, and early in the course of the disease there is an increase in one or more of the formed myeloid elements (granulocytes, platelets, and/or red cells) in the peripheral blood. Differentiation is initially unaffected or altered in relatively subtle ways, such that patients tend to suffer from symptoms related to an increase in myeloid progenitors and formed elements. Some myeloproliferative disorders are associated with striking increases in basophil counts (basophilia) or eosinophil counts (eosinophilia), features that can serve as important diagnostic clues.
Unfortunately, as with virtually all cancers, the myeloproliferative disorders are prone to acquire additional mutations during their course that lead to more aggressive behavior and a worsening of the clinical picture. Disease progression can take one of two forms: 1) transformation to acute leukemia, in which the marrow is replaced by undifferentiated cells called blasts, and 2) progression to myelofibrosis, in which the marrow is replaced by collagen deposited by reactive fibroblasts. Both of these events are understandably associated with the development of low blood counts (cytopenias) due to marrow dysfunction.
Diagnosis of the major subtypes of myeloproliferative disorders is based on the detection of mutations in specific tyrosine kinases combined with clinical features and laboratory findings (summarized in Table 20-1). The myeloproliferative disorders are not common, but they are of more than passing interest because an understanding of their pathogenesis has led to the development of effective, relatively nontoxic therapies that specifically target the underlying causative molecular lesions. In the section that follows, we describe the important myeloproliferative disorders.
| Entity | Cell of Origin | Tyrosine Kinase Mutation | Typical Peripheral Blood Findings | Clinical Features |
|---|---|---|---|---|
| Chronic myelogenous leukemia | Hematopoietic stem cell | BCR-ABL fusion gene | Neutrophilia, eosinophilia, basophilia, left-shift, thrombocytosis, mild anemia | Symptoms related to extensive extramedullary hematopoiesis; transformation to acute leukemia in 100% of cases; infrequent progression to myelofibrosis |
| Polycythemia vera | Early myeloid progenitor | JAK2 point mutations | Pancytosis (erythrocytosis, neutrophilia, basophilia, and thrombocytosis) | Symptoms related to high red cell counts; rarely transforms to acute leukemia; progresses to myelofibrosis in 10%-20% of cases |
| Essential thrombocytosis | Early myeloid progenitor | JAK2 or MPL point mutations | Thrombocytosis | Symptoms related to high platelet counts; rarely transforms to acute leukemia; progresses to myelofibrosis in 10%-20% of cases |
| Primary myelofibrosis | Early myeloid progenitor | JAK2 or MPL point mutations | Anemia, leukoerythroblastosis | Symptoms related to early-onset myelofibrosis, which leads to anemia and extensive extramedullary hematopoiesis; rarely transforms to acute leukemia |
| Chronic eosinophilic leukemia | Early myeloid progenitor or hematopoietic stem cell | PDGFR-α or PDGFR-β fusion genes | Leukocytosis, eosinophilia, monocytosis (particularly with PDGFR-β rearrangements) | Symptoms related to fibrosis of tissues infiltrated by eosinophils, including the heart and skin |
| Systemic mastocytosis | Early myeloid progenitor | c-KIT point mutations | None | Symptoms related to mediators released from mast cells, including pruritus, diarrhea, flushing, diaphoresis, syncope, and osteopenia |
| Myeloproliferative disorder associated with FGFR1 rearrangement | Hematopoietic stem cell | Rearrangements of FGFR1 (usually translocations) | Leukocytosis, eosinophilia | Initial symptoms related to myeloproliferative disease; however, usually progresses to acute lymphoblastic leukemia/lymphoblastic lymphoma; may recur after therapy as acute myeloid leukemia. |
CHRONIC MYELOGENOUS LEUKEMIA
Chronic myelogenous leukemia (CML) is always associated with a somatic mutation that produces a BCR-ABL fusion gene, usually a balanced translocation between chromosomes 9 and 22 (the so-called Philadelphia chromosome; see Chapter 19). The fusion gene encodes a chimeric BCR-ABL tyrosine kinase that self-associates through its BCR moiety, leading to constitutive activation of the ABL kinase. In most instances, the positions of the chromosomal breakpoints are such that the BCR-ABL protein is 210 kDa in size. The cause of the translocation is not known. The incidence of CML was increased in survivors of the atomic bombs in Nagasaki and Hiroshima, possibly indicating that radiation-induced DNA damage can promote the formation of the 9-22 translocation, or t(9;22). However, very sensitive polymerase chain reaction (PCR) assays have enabled detection of low levels of BCR-ABL fusion transcripts in a high percentage of normal blood donors, the vast majority of whom will never develop CML. It therefore appears that the BCR-ABL fusion gene is necessary but not sufficient for CML development.
The BCR-ABL kinase activates the RAS, JAK-STAT, and PI-3 kinase/AKT pathways, the same pathways that are stimulated by hematopoietic growth -factors in normal progenitors. As a result, there is an overgrowth of granulocytic and megakaryocytic precursors in the bone marrow (Fig. 20-1). For unclear reasons, erythroid elements are not as responsive to the effects of the BCR-ABL kinase, and, as a result, the ratio of granulocytic to erythroid progenitors in the marrow is markedly elevated. Seeding of neoplastic hematopoietic stem cells and marrow progenitors to the spleen and liver results in extramedullary hematopoiesis, which often produces organomegaly (Fig. 20-2). The peripheral blood smear typically shows neutrophilia with a left-shift (the presence of immature granulocytic forms) and varying degrees of eosinophilia, basophilia, monocytosis, and thrombocytosis plus mild anemia (Fig. 20-3).
FIGURE 20-1
Chronic myelogenous leukemia (bone marrow biopsy). A) The biopsy is hypercellular (no fat) due to the presence of excess granulocytes and granulocytic precursors, including eosinophilic forms, and increased numbers of megakaryocytes. Compare its appearance to a normal bone marrow biopsy B), which is roughly 50% cellular and contains normal proportions of cells from the erythroid, myeloid, and megakaryocytic lineages.
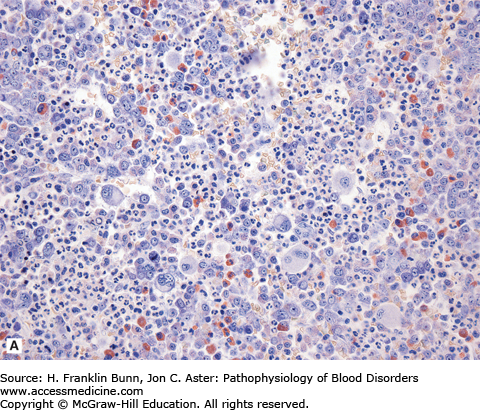
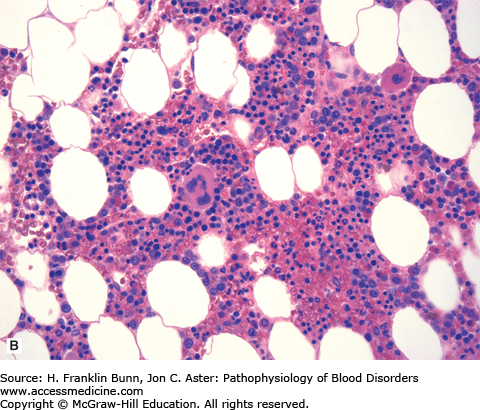
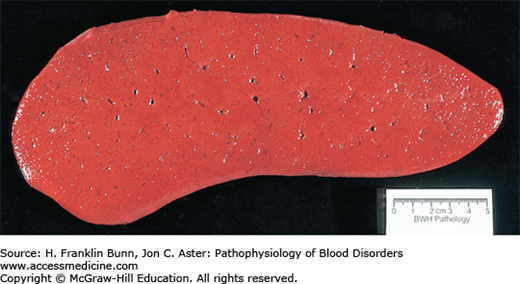
FIGURE 20-3
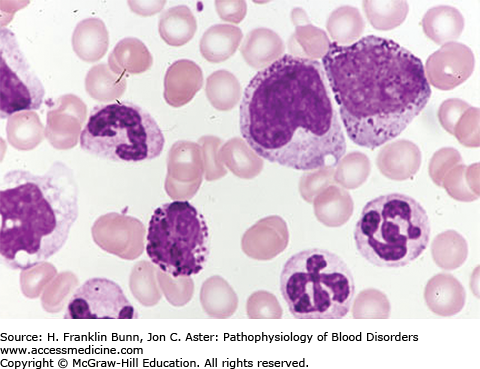
Chronic myelogenous leukemia, peripheral blood. Note the leukocytosis, the presence of immature granulocytic precursors (promyelocytes), the abnormal nuclear segmentation of the neutrophils, and the presence of a basophil, all of which are characteristic. (Courtesy of Lichtman MA, Shafer MS, Felgar RE, Wang N, Lichtman’s Atlas of Hematology ([Online via Access Medicine], McGraw-Hill.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


