CHAPTER 39 Muscles
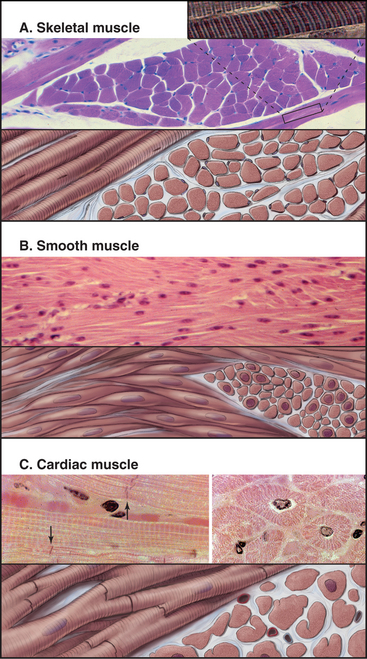
Vertebrates have three types of specialized contractile cells—smooth muscle, skeletal muscle, and cardiac muscle—that use actin and myosin to generate powerful, unidirectional movements (Fig. 39-1). These muscles have much in common but differ in their activation mechanisms, arrangement of contractile filaments, and energy supplies. This provides three options for physiological responses. The nervous system controls the timing, force, and speed of skeletal muscle contraction over a wide range. Cardiac muscle generates its own rhythmic, fatigue-free contractions that spread through the heart in a highly reproducible fashion. Neurotransmitters, acting like hormones, regulate the force and frequency of heartbeats over a narrow range. Nerves, hormones, and intrinsic signals control the activity of smooth muscles, which contract slowly but maintain tension very efficiently.
Skeletal Muscle
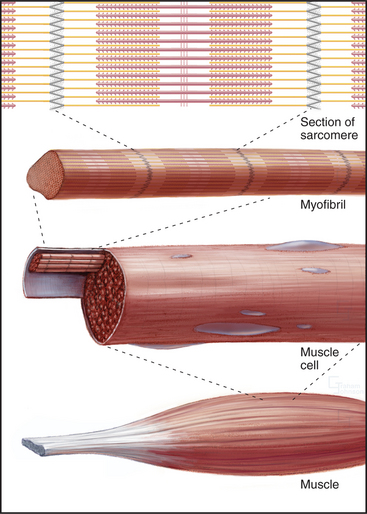
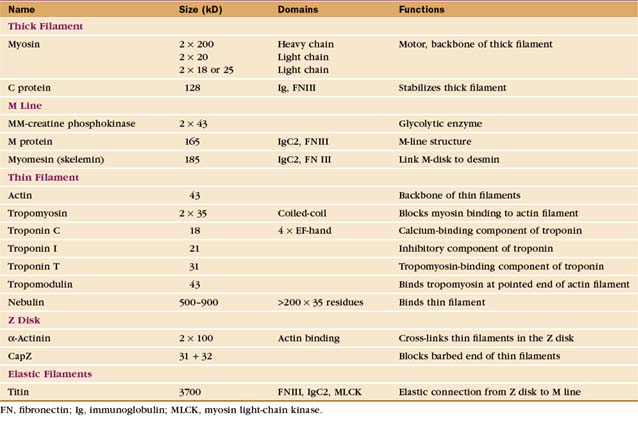
Skeletal muscle cells are optimized for rapid, forceful contractions. Accordingly, they have a massive concentration of highly ordered contractile units composed of actin, myosin, and associated proteins (Fig. 39-2). Actin and myosin filaments are organized into sarcomeres, aligned contractile units that give the cells a striped appearance in the microscope. For this reason, they are called striated muscles. Myosin uses ATP hydrolysis to power contraction, which results from myosin-powered sliding of actin-based thin filaments past myosin-containing thick filaments. Speed is achieved by linking many sarcomeres in series. Force is determined by the number of sarcomeres contracting in parallel.
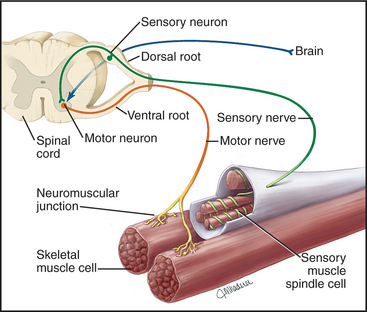
Although skeletal muscle cells have only two states—inactive (relaxed) or active (contracting)—skeletal muscles produce a wide range of contractions, varying from slow and delicate to rapid and forceful. These graded contractions are achieved by varying the number of muscle cells activated by voluntary or reflex signals from the nervous system (Fig. 39-14). Nerve impulses stimulate a transient rise in cytoplasmic calcium that activates the contractile proteins.
Organization of the Skeletal Muscle Contractile Apparatus
Skeletal muscle cells (also called muscle fibers in the physiological literature) are among the largest cells of vertebrates. During development, mesenchymal stem cells give rise to progenitor cells with single nuclei called myoblasts. A family of master transcription factors, including MyoD and myogenin, coordinates the expression of specialized muscle proteins. As they differentiate, numbers of myoblasts fuse and elongate to form muscle cells with multiple nuclei and lengths of millimeters to tens of centimeters. The number of muscle cells is determined genetically and is relatively stable throughout life even as the size of the cells varies with the level of exercise and nutrition. Mature muscles harbor small numbers of stem cells (called satellite cells [see Fig. 41-15]) with a limited capacity to differentiate and repair damage.
A basal lamina (see Fig. 29-18C) surrounds and supports each muscle cell. At the ends of each cell, actin thin filaments are anchored to the plasma membrane at myotendinous junctions, which are similar to focal contacts (see Fig. 30-11). Integrins spanning the membrane link actin filaments to the basal lamina and to collagen fibrils of tendons. These physical connections transmit contractile force to the skeleton.
Organization of the Actomyosin Apparatus
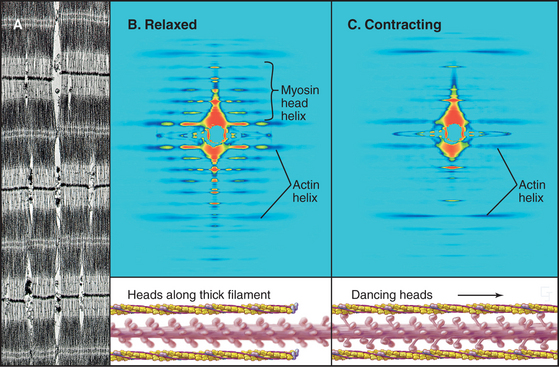
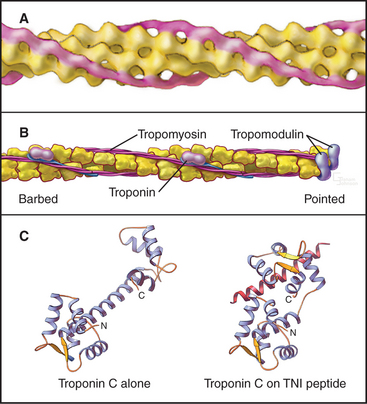
Interdigitation of thick, bipolar, myosin filaments and thin actin filaments in the sarcomeres of living muscle cells is so precise (Fig. 39-3) that it yields an X-ray diffraction pattern (Fig. 39-13) revealing the spacing of the filaments and the helical repeats of their subunits to a resolution of about 3 nm. Z disks at both ends of the sarcomere anchor the barbed ends of the actin filaments, so their pointed ends are near the center of the sarcomere. Myosin heads project from the surface of thick filaments, whereas their tails are anchored in the filament backbone. Thick and thin filaments overlap, with the myosin heads only a few nanometers away from adjacent actin filaments. The alignment and interdigitation of the filaments facilitate the sliding interactions required to produce contraction.

An important, simplifying architectural feature is that sarcomeres are symmetrical about their middles (Fig. 39-3). Consequently, the polarity of myosin relative to the actin filaments is the same in both halves of the sarcomere, allowing the same force-generating mechanism to work at both ends of the bipolar myosin filaments. Sarcomeres are organized end to end into long, cylindrical assemblies called myofibrils (Fig. 39-2) that retain their contractility even after isolation from muscle.
Thin Filaments
Thin filaments consist of actin and the tightly bound regulatory proteins troponin and tropomyosin (Fig. 39-4). When the concentration of Ca2+ is low in cytoplasm, troponin and tropomyosin inhibit the actin-activated ATPase of myosin. Tropomyosin, a 40-nm long coiled-coil of two α-helical polypeptides (see Fig. 3-10), binds laterally to seven contiguous actin subunits as well as head to tail to neighboring tropomyosins, forming a continuous strand along the whole thin filament. Troponin (TN) consists of three different subunits called TNC, TNI, and TNT (Table 39-1). TNT anchors troponin to tropomyosin. Like calmodulin (see Fig. 3-12 and Chapter 26), TNC is a dumbbell-shaped protein with four EF-hand motifs to bind divalent cations. In resting muscle the C-terminal globular domain of TNC binds two Mg2+ ions and an N-terminal α-helix of TNI, while the two low-affinity sites in the N-terminal globular domain of TNC are empty. Ca2+ binding to the low-affinity sites during muscle activation exposes a new binding site for TNI. The resulting conformational change in TNI allows tropomyosin to expose myosin-binding sites on the actin filament.
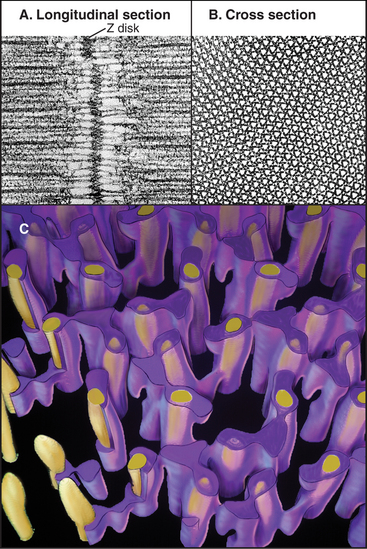
A protein meshwork in the Z disk anchors the barbed end of each thin filament (Fig. 39-5). Some cross-links between actin filaments consist of α-actinin, a short rod with actin-binding sites on each end (see Fig. 33-16). At least a half dozen structural proteins stabilize the Z disk through interactions with α-actinin, actin, and titin in the Z disk.
Proteins cap both ends of thin filaments. Cap-Z, the muscle isoform of capping protein (see Fig. 33-14), binds the barbed ends of thin filaments with high affinity, limiting actin subunit addition or loss. Tropomodulin associates with both tropomyosin and actin to cap and stabilize the pointed end of thin filaments (Fig. 39-4B).
Tropomyosin and a gigantic filamentous protein, nebulin, stabilize thin filaments laterally. Nebulin consists of about 185 imperfect repeats of 35 amino acids that interact with each actin subunit, tropomyosin, and troponin along the length of thin filaments. Interactions with tropomodulin and Z disk proteins anchor nebulin at the two ends of the thin filament. Acting as a ruler and a cap, nebulin and tropomodulin help to set the length of thin filaments.
Thick Filaments
The self-assembly of myosin II (see Fig. 5-7) establishes the bipolar architecture of striated muscle thick filaments (Fig. 39-6). Some features of thick filaments are invariant, such as a backbone consisting of myosin tails, a surface array of myosin heads, the 14.3-nm stagger between rows of heads, and a central bare zone formed by antiparallel packing of tails. Filaments may vary in length, diameter, and organization of the helical array of heads in various species. Invertebrate thick filaments have a core of paramyosin, a second coiled-coil protein, which is not found in vertebrates.
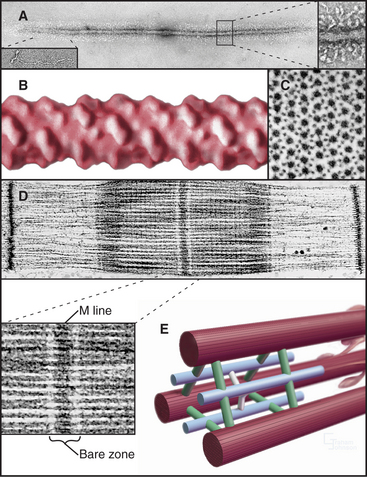
Several accessory proteins stabilize thick filaments (Table 39-1). Thick filaments in most striated muscles are girdled at intervals by semicircular bands of a protein that is, coincidentally, called C-protein. C-protein consists of fibronectin III and immunoglobulin domains. The “M line” in the center of the sarcomere is a three-dimensional array of protein cross-links that maintains the precise registration of thick filaments. At least three structural proteins and the enzyme MM-creatine phosphokinase (which transfers phosphate from creatine-phosphate to ADP) are located in the M line.
Titin Filaments
A third array of protein filaments lies parallel to the thin and thick filaments, connecting the Z disk to the thick filaments and the M line (Fig. 39-7). Hard to preserve for electron microscopy, these diaphanous filaments were neglected for years. Each filament is a single polypeptide named titin (after mythological giants), so named because of its remarkable size: more than 30,000 amino acids folded into a linear array of 300 immunoglobulin and fibronectin II domains measuring more than 1.2 μm long. Titin is thought to be the largest protein encoded by the human genome.
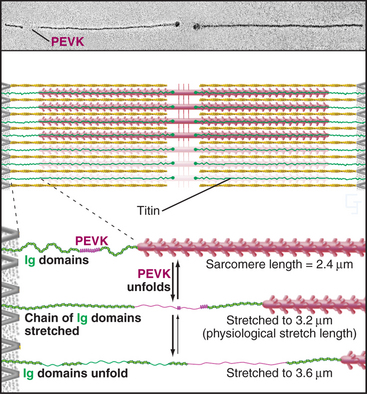
Titin molecules are elastic, and this accounts for the passive resistance to stretching of relaxed muscle. Their connections to the Z disk and thick filaments provide elastic continuity from one sarcomere to the next and keep the thick filaments centered in the sarcomere during contraction. If titin molecules are broken experimentally, thick filaments slide out of register toward one Z disk during contraction. Two features provide the elasticity during short (∼0.3 μm per titin), physiological stretches: The irregular chain of immunoglobulin domains in the I band straightens out, and a segment of the polypeptide rich in proline, glutamic acid, valine, and lysine (the PEVK domain) stretches. Stretching decreases entropy and provides the energy for elastic recoil. (See Fig. 29-12 for another example of an entropic spring in biology.) Extreme stretching unfolds Ig domains one by one.
Intermediate Filaments
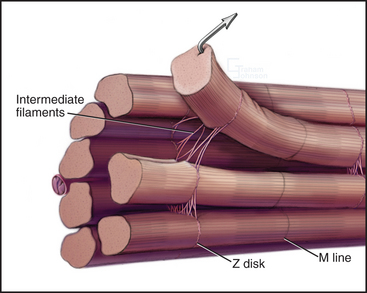
Desmin intermediate filaments (see Chapter 35) help to align the sarcomeres laterally (Fig. 39-8) by linking each Z disk to its neighbors and to specialized attachment sites on the plasma membrane. Myofibrils near the cell surface are attached to the plasma membrane at specializations called costameres. In addition to desmin, costameres contain several cytoskeletal proteins (vinculin, talin, spectrin, and ankyrin) found in focal contacts and adherens junctions of nonmuscle cells (see Figs. 30-11 and 31-7). Desmin mutations in humans cause disorganization of myofibrils, resulting in generalized muscle failure.
Organization of the Muscle Membrane System
Structural Proteins of the Plasma Membrane: Defects in Muscular Dystrophies
In addition to providing a permeability barrier, the plasma membrane of the muscle cell must maintain its integrity while being subjected to years of forceful contractions. Accordingly, the membrane is stabilized by a transmembrane complex of proteins, the dystroglycan-sarcoglycan complex, that links the internal membrane skeleton to the basal lamina outside (Fig. 39-9 and Table 39-2). Occasional breaches of the membrane are inevitable, so muscle cells also depend on a repair process that reseals holes. If membrane damage exceeds the repair capacity, muscle cells degenerate locally (segmental necrosis) or globally. Cell death beyond the ability of muscle stem cells to repair the tissue results in muscular dystrophy. The age of onset and clinical features of inherited muscular dystrophies depend on the molecular defect. Patients with severe defects develop progressive muscle weakness as children. Ultimately, failure of respiratory muscles is fatal.
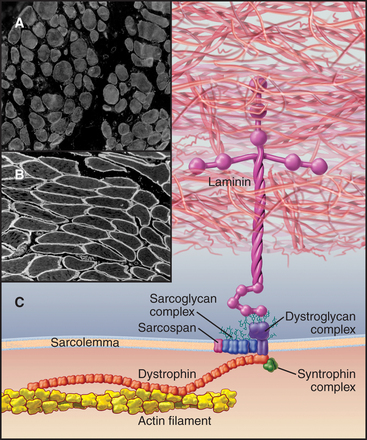
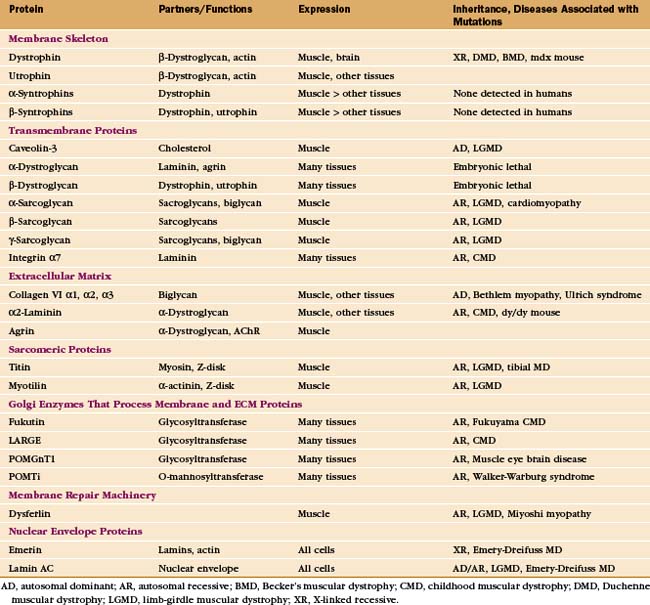
Mutations in more than 40 human genes have been linked to muscular dystrophies (Table 39-2), so it is possible that the malfunction or lack of any molecule in the system that maintains the integrity of the plasma membrane will cause disease. Disease-causing mutations occur in genes for the proteins of the dystroglycan-sarcoglycan complex, extracellular matrix proteins, Golgi apparatus enzymes that process these structural proteins, and the membrane repair machinery. Some of these mutations also affect the nervous system. Mutations are usually autosomal-recessive and are not uncommon. About one in several thousand humans develops some form of muscular dystrophy, because they inherit mutations in both copies of one of the sensitive genes. The mechanism of disease in muscular dystrophies is similar to that in hereditary spherocytosis, in which deficiencies of the membrane skeleton make red blood cells susceptible to mechanical damage (see Fig. 6-10).
The proteins that stabilize muscle membranes escaped detection until the late 1980s, when x-linked mutations in the dystrophin gene were discovered to cause Duchenne’s muscular dystrophy, the most common human form of the disease. Dystrophin is an enormous member of the α-actinin superfamily of actin-binding proteins (see Fig. 33-16). The dystroglycan-sarcoglycan complex was found when it copurified with dystrophin after solubilizing the membrane with detergents. Loss of any protein of the dystrophin-dystroglycan-sarcoglycan complex typically leads to secondary loss of the other proteins from muscles.
Dystroglycans and a dystrophin homolog, utrophin, participate in clustering acetylcholine receptors at the neuromuscular junction, the chemical synapse between motor neurons and skeletal muscle (see Fig. 11-8). When, during development, a motor neuron arrives at the surface of its target muscle cell, the neuron secretes an adhesive protein called agrin, which is incorporated into the basal lamina, immediately adjacent to the nerve terminal. Dystroglycan binds agrin and positions associated acetylcholine receptors at the site where they receive acetylcholine secreted by the nerve in response to an action potential.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


