] embodies the properties that typify all muscarinic agonists and will serve as our prototype for the group.
Mechanism of Action
Bethanechol is a direct-acting muscarinic agonist. The drug binds reversibly to muscarinic cholinergic receptors to cause activation. At therapeutic doses, bethanechol acts selectively at muscarinic receptors, having little or no effect on nicotinic receptors, either in ganglia or in skeletal muscle.
Pharmacologic Effects
Bethanechol can elicit all of the responses typical of muscarinic receptor activation. Accordingly, we can readily predict the effects of bethanechol by knowing the information on muscarinic responses summarized in Table 11.2.
The principal structures affected by muscarinic activation are the heart, exocrine glands, smooth muscles, and eyes. Muscarinic agonists act on the heart to cause bradycardia (decreased heart rate) and on exocrine glands to increase sweating, salivation, bronchial secretions, and secretion of gastric acid. In smooth muscles of the lungs and GI tract, muscarinic agonists promote contraction. The result is constriction of the bronchi and increased tone and motility of GI smooth muscle. In the bladder, muscarinic activation causes contraction of the detrusor muscle and relaxation of the trigone and sphincter; the result is bladder emptying. In vascular smooth muscle, these drugs cause relaxation; the resultant vasodilation can produce hypotension. Activation of muscarinic receptors in the eyes has two effects: (1) miosis (pupillary constriction), and (2) contraction of the ciliary muscle, resulting in accommodation for near vision. (The ciliary muscle, which is attached to the lens, focuses the eyes for near vision by altering lens curvature.)
Pharmacokinetics
Bethanechol is available for oral administration. Effects begin in 30 to 60 minutes and persist for about 1 hour. Because bethanechol is a quaternary ammonium compound (Fig. 12.1), the drug crosses membranes poorly. As a result, only a small fraction of each dose is absorbed.

Preparations, Dosage, and Administration
Preparation and dosing of bethanechol and other cholinesterase inhibitors is provided in Table 12.2. Administration guidelines are also provided.
TABLE 12.2
Preparation, Dosage, and Administration of Muscarinic Agonists
| Drug Class or Drug | Preparation | Dosage | Administration |
| MUSCARINIC AGONISTS | |||
Bethanechol [Urecholine, Duvoid  ] ] | Tablets: 5, 10, 25, and 50 mg | 10–50 mg 3–4 times a day | 1 hour before meals or 2 hours after to prevent nausea and vomiting |
| Cevimeline [Evoxac] | Capsules: 30 mg | 30 mg 3 times a day | May be given without regard to food. Food decreases the rate of absorption but not the amount absorbed. |
Pilocarpine ophthalmic [Isopto Carpine, Diocarpine  , Pilopine HS] , Pilopine HS] | Solution: 1% in 15 mL, 2% in 15 mL, & 4% in 15 mL Gel: 4% | Solution: 1–2 drops to affected eye up to 6 times a day Gel: apply a 0.5-inch ribbon onto the lower conjunctival sac at bedtime | Apply pressure to lacrimal area for 1–2 minutes after administration. If both solution and gel are needed, patient should apply the solution first and wait 5 minutes before applying the gel. |
| Pilocarpine systemic [Salagen] | Tablets: 5 mg, 7.5 mg | Sjögren syndrome: 5 mg 4 times a day Postradiotherapy for cancer: 5 mg 3 times a day initially; may be titrated upward to 10 mg 3 times a day | Avoid administration with high-fat meals due to decreased rate of absorption. |
Therapeutic Uses
Although bethanechol can produce a broad spectrum of pharmacologic effects, the drug is approved only for urinary retention.
Urinary Retention.
Bethanechol relieves urinary retention by activating muscarinic receptors of the urinary tract. Muscarinic activation relaxes the trigone and sphincter muscles and increases voiding pressure by contracting the detrusor muscle, which composes the bladder wall. It is approved to treat urinary retention in postoperative and postpartum patients and to treat retention secondary to neurogenic atony of the bladder. The drug should not be used to treat urinary retention caused by physical obstruction of the urinary tract because increased pressure in the tract in the presence of blockage could cause injury.
GI Uses.
Bethanechol has been used off-label to treat gastroesophageal reflux. Benefits may result from increased esophageal motility and increased pressure in the lower esophageal sphincter.
Bethanechol can help treat disorders associated with GI paralysis. Benefits derive from increased tone and motility of GI smooth muscle. Specific applications are adynamic ileus, gastric atony, and postoperative abdominal distention. Bethanechol should not be given if physical obstruction of the GI tract is present because, in the presence of blockage, increased propulsive contractions might result in damage to the intestinal wall.
Adverse Effects
In theory, bethanechol can produce the full range of muscarinic responses as side effects. However, with oral dosing, side effects are relatively rare.
Cardiovascular System.
Bethanechol can cause hypotension (secondary to vasodilation) and bradycardia. Accordingly, the drug is contraindicated for patients with low blood pressure or low cardiac output.
GI System.
At usual therapeutic doses, bethanechol can cause excessive salivation, increased secretion of gastric acid, abdominal cramps, and diarrhea. Higher doses can cause involuntary defecation. Bethanechol is contraindicated in patients with gastric ulcers because stimulation of acid secretion could intensify gastric erosion, causing bleeding and possibly perforation. The drug is also contraindicated for patients with intestinal obstruction and for those recovering from recent surgery of the bowel. In both cases, the ability of bethanechol to increase the tone and motility of intestinal smooth muscle could result in rupture of the bowel wall.
Urinary Tract.
Because of its ability to contract the bladder detrusor and thereby increase pressure within the urinary tract, bethanechol can be hazardous to patients with urinary tract obstruction or weakness of the bladder wall. In both groups, elevation of pressure within the urinary tract could rupture the bladder. Accordingly, bethanechol is contraindicated for patients with either disorder.
Exacerbation of Asthma.
By activating muscarinic receptors in the lungs, bethanechol can cause bronchoconstriction. Accordingly, the drug is contraindicated for patients with latent or active asthma. Of course, it stands to reason that muscarinic agonists may also complicate other respiratory disorders.
Dysrhythmias in Hyperthyroid Patients.
Bethanechol is contraindicated for people with hyperthyroidism. If given to patients with this condition, bethanechol may increase heart rate to the point of initiating a dysrhythmia. (Note that increased heart rate is opposite to the effect that muscarinic agonists have in most patients.) The mechanism of dysrhythmia induction is explained here.
When hyperthyroid patients are given bethanechol, their initial cardiovascular responses are like those of anyone else: bradycardia and hypotension. In reaction to hypotension, the baroreceptor reflex attempts to return blood pressure to normal. Part of this reflex involves the release of norepinephrine from sympathetic nerves that regulate heart rate. In patients who are not hyperthyroid, norepinephrine release serves to increase cardiac output and thus helps restore blood pressure. However, in hyperthyroid patients, norepinephrine can induce cardiac dysrhythmias. The reason for this unusual response is that, in hyperthyroid patients, the heart is exquisitely sensitive to the effects of norepinephrine, and hence relatively small amounts can cause stimulation sufficient to elicit a dysrhythmia.
Other Muscarinic Agonists
Cevimeline
Actions and Uses
Cevimeline [Evoxac] is a derivative of acetylcholine with actions much like those of bethanechol. The drug is indicated for relief of xerostomia (dry mouth) in patients with Sjögren syndrome, an autoimmune disorder characterized by xerostomia. It has also been used to manage keratoconjunctivitis sicca (inflammation of the cornea and conjunctiva) and dry eye. Dry mouth, left untreated, can lead to multiple complications, including periodontal disease, dental caries, altered taste, oral ulcers and candidiasis, and difficulty eating and speaking. Cevimeline relieves dry mouth by activating muscarinic receptors on residual healthy tissue in salivary glands, thereby promoting salivation. Because it stimulates salivation, cevimeline may also benefit patients with xerostomia induced by radiation therapy for head and neck cancer, although the drug is not approved for this use. The drug also increases tear production, which can help relieve keratoconjunctivitis and dry eye.
Adverse Effects
Adverse effects result from activating muscarinic receptors and hence are similar to those of bethanechol. The most common effects are excessive sweating, nausea, rhinitis, and diarrhea. To compensate for fluid loss caused by sweating and diarrhea, patients should increase fluid intake. Like bethanechol, cevimeline promotes miosis (constriction of the pupil) and may also cause blurred vision. Both actions can make driving dangerous, especially at night.
Activation of cardiac muscarinic receptors can reduce heart rate and slow cardiac conduction. Accordingly, cevimeline should be used with caution in patients with a history of heart disease.
Because muscarinic activation increases airway resistance, cevimeline is contraindicated for patients with uncontrolled asthma and should be used with caution in patients with controlled asthma, chronic bronchitis, or chronic obstructive pulmonary disease (COPD). Cevimeline is also contraindicated for people with both narrow-angle glaucoma and iritis.
Drug Interactions
Cevimeline can intensify cardiac depression caused by beta blockers because both drugs decrease heart rate and cardiac conduction.
Beneficial effects of cevimeline can be antagonized by drugs that block muscarinic receptors. Among these are atropine, tricyclic antidepressants (e.g., imipramine), antihistamines (e.g., diphenhydramine), and phenothiazine antipsychotics (e.g., chlorpromazine).
Pilocarpine
Pilocarpine is a muscarinic agonist used mainly for topical therapy of glaucoma, an ophthalmic disorder characterized by elevated intraocular pressure (IOP) with subsequent injury to the optic nerve. The basic pharmacology of pilocarpine and its use in glaucoma are discussed in Chapter 84.
In addition to its use in glaucoma, oral pilocarpine is approved for treatment of dry mouth resulting from Sjögren syndrome or from salivary gland damage caused by radiation therapy of head and neck cancer. For these applications, pilocarpine is available under the trade name Salagen. It may also be given to manage dry mouth secondary to head and neck cancer. At lower doses, the principal adverse effect is sweating. However, if dosage is excessive, pilocarpine can produce the full spectrum of muscarinic effects.
Cholinesterase Inhibitors
Cholinesterase inhibitors are drugs that prevent the degradation of acetylcholine by acetylcholinesterase (also known simply as cholinesterase). Cholinesterase inhibitors are also known as anticholinesterase drugs. By preventing the breakdown of acetylcholine, cholinesterase inhibitors increase the amount of acetylcholine available to activate receptors, thus enhancing cholinergic action. Because cholinesterase inhibitors do not bind directly with cholinergic receptors, they are viewed as indirect-acting cholinergic agonists. Use of cholinesterase inhibitors results in transmission at all cholinergic junctions (muscarinic, ganglionic, and neuromuscular), so these drugs can elicit a broad spectrum of responses. Because they lack selectivity, cholinesterase inhibitors have limited therapeutic applications.
There are two basic categories of cholinesterase inhibitors: (1) reversible inhibitors and (2) irreversible inhibitors. The reversible inhibitors produce effects of moderate duration, and the irreversible inhibitors produce effects of long duration.
Reversible Cholinesterase Inhibitors
Neostigmine
Neostigmine [Bloxiverz, Prostigmin] typifies the reversible cholinesterase inhibitors and will serve as our prototype for the group. The principal indication of Prostigmin is management of myasthenia gravis (MG). Bloxiverz is used to reverse the actions of nondepolarizing neuromuscular blockade after surgery; however, this use is beyond the scope of this book.
Chemistry.
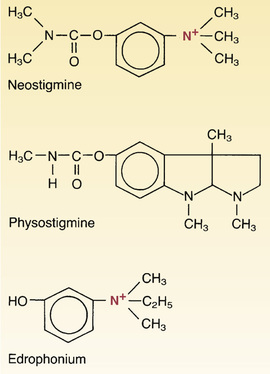
As shown in Fig. 12.2, neostigmine contains a quaternary nitrogen atom and hence always carries a positive charge. Because of this charge, neostigmine cannot readily cross membranes, including those of the GI tract, blood-brain barrier, and placenta. Consequently, neostigmine is absorbed poorly after oral administration and has minimal effects on the brain and fetus.
Mechanism of Action.
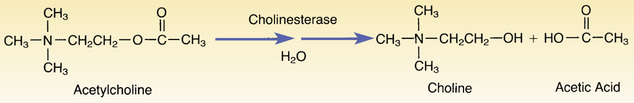
Neostigmine and the other reversible cholinesterase inhibitors act as substrates for cholinesterase. As indicated in Fig. 12.2, the normal function of cholinesterase is to break down acetylcholine into choline and acetic acid. (This process is called a hydrolysis reaction because of the water molecule involved.) The overall reaction between acetylcholine and cholinesterase is extremely fast. As a result, one molecule of cholinesterase can break down a huge amount of acetylcholine in a very short time.
The reaction between neostigmine and cholinesterase is much like the reaction between acetylcholine and cholinesterase. The only difference is that cholinesterase splits neostigmine more slowly than it splits acetylcholine. Hence, after neostigmine becomes bound to cholinesterase, the drug remains in place for a relatively long time. Because cholinesterase remains bound until it finally succeeds in degrading neostigmine, less cholinesterase is available to catalyze the breakdown of acetylcholine. As a result, more acetylcholine is available to activate cholinergic receptors.
Pharmacologic Effects.
By decreasing breakdown of acetylcholine, neostigmine and the other cholinesterase inhibitors make more acetylcholine available, and this can intensify transmission at virtually all junctions where acetylcholine is the transmitter. In sufficient doses, cholinesterase inhibitors can produce skeletal muscle stimulation, ganglionic stimulation, activation of peripheral muscarinic receptors, and activation of cholinergic receptors in the central nervous system (CNS). However, when used therapeutically, cholinesterase inhibitors usually affect only muscarinic receptors on organs and nicotinic receptors of the neuromuscular junction (NMJ). Ganglionic transmission and CNS function are usually unaltered.
Muscarinic Responses.
Muscarinic effects of the cholinesterase inhibitors are identical to those of the direct-acting muscarinic agonists. By preventing breakdown of acetylcholine, cholinesterase inhibitors can cause bradycardia, bronchial constriction, urinary urgency, increased glandular secretions, increased tone and motility of GI smooth muscle, miosis, and focusing of the lens for near vision.
Neuromuscular Effects.
The effects of cholinesterase inhibitors on skeletal muscle are dose dependent. At therapeutic doses, these drugs increase force of contraction. In contrast, toxic doses reduce force of contraction. Contractile force is reduced because excessive amounts of acetylcholine at the NMJ keep the motor end plate in a state of constant depolarization, causing depolarizing neuromuscular blockade.
Central Nervous System.
Effects on the CNS vary with drug concentration. Therapeutic levels can produce mild stimulation, whereas toxic levels depress the CNS, including the areas that regulate respiration. However, keep in mind that, for CNS effects to occur, the inhibitor must first penetrate the blood-brain barrier, which some cholinesterase inhibitors can do only when present in very high concentrations.
Pharmacokinetics.
Neostigmine may be administered by intramuscular (IM), intravenous (IV), or subcutaneous (subQ) injection. Because neostigmine carries a positive charge, the drug is poorly absorbed after oral administration; hence oral formulations have been discontinued in the United States, although they remain available in some other countries. After it is absorbed, neostigmine can reach sites of action at the NMJ and peripheral muscarinic receptors, but cannot cross the blood-brain barrier to affect the CNS. Duration of action is 2 to 4 hours. Neostigmine is eliminated by enzymatic degradation by cholinesterase.
Preparation, Dosage, and Administration.
Preparation and dosage of neostigmine and other cholinesterase inhibitors are provided in Table 12.3. Administration guidelines are also provided.
TABLE 12.3
Preparation, Dosage, and Administration of Cholinesterase Inhibitors
| Drug Class or Drug | Preparation | Dosage | Administration |
| CHOLINESTERASE INHIBITORS | |||
| Neostigmine [Prostigmin] | Prostigmin: 0.5-mg/mL solution available in 1-mL and 10-mL vials Generic: 0.5 mg/mL and 1 mg/mL, both in 10-mL vials | Myasthenia gravis treatment (highly individualized): 0.5 mg IM or subQ initially with additional dosing based on patient response. Typical dosing is 15–375 mg/day in divided doses | Timing administration so that peak effects occur at meal time may help with eating and swallowing |
| Physostigmine | 1 mg/mL in 2-mL vials | Reversal of anticholinergic toxicity: adults—0.5–2 mg IM or IV Children: 0.02 mg/kg Dose may be repeated every 10–30 minutes as needed | Rapid IV administration can cause respiratory distress, bradycardia, and seizures. Limit rate to 1 mg/min in adults or 0.5 mg/min in children. |
Pyridostigmine [Mestinon Regonol, Mestinon-SR  ] ] | Syrup: 60 mg/5 mL Tablet IR: 60 mg Tablet ER: 180 mg | Myasthenia gravis treatment (highly individualized) IR: 60–1,500 mg/day Typical dosing 600 mg/day divided into 5 doses ER: 180–540 mg once or twice a day (It may be necessary to use both IR and ER dosing to sustain effects) | Take the ER tablets whole. |
Adverse Effects
Excessive Muscarinic Stimulation.
Accumulation of acetylcholine at muscarinic receptors can result in excessive salivation, increased gastric secretions, increased tone and motility of the GI tract, urinary urgency, bradycardia, sweating, miosis, and spasm of accommodation (focusing of the lens for near vision). If necessary, these responses can be suppressed with atropine.
Neuromuscular Blockade.
If administered in toxic doses, cholinesterase inhibitors can cause accumulation of acetylcholine in amounts sufficient to produce depolarizing neuromuscular blockade. Paralysis of the respiratory muscles can be fatal.
Precautions and Contraindications.
Most of the precautions and contraindications regarding the cholinesterase inhibitors are the same as those for the direct-acting muscarinic agonists. These include obstruction of the GI tract, obstruction of the urinary tract, peptic ulcer disease, asthma, coronary insufficiency, and hyperthyroidism.
Drug Interactions
The effects of cholinesterase inhibitors at muscarinic receptors are opposite to those of atropine (and all other muscarinic antagonists). Consequently, cholinesterase inhibitors can be used to overcome excessive muscarinic blockade caused by atropine. Conversely, atropine can be used to reduce excessive muscarinic stimulation caused by cholinesterase inhibitors.
Other Reversible Cholinesterase Inhibitors
Physostigmine.
The basic pharmacology of physostigmine is identical to that of neostigmine. In contrast to neostigmine, physostigmine is not a quaternary ammonium compound and hence does not carry a charge. Because physostigmine is uncharged, physostigmine readily crosses membranes, whereas neostigmine does not.
Physostigmine is the drug of choice for treating poisoning by atropine and other drugs that cause muscarinic blockade, including antihistamines and phenothiazine antipsychotics—but not tricyclic antidepressants, owing to a risk for causing seizures and cardiotoxicity. Physostigmine counteracts antimuscarinic poisoning by causing acetylcholine to build up at muscarinic junctions. The accumulated acetylcholine competes with the muscarinic blocker for receptor binding and thereby reverses receptor blockade. Physostigmine is preferred to neostigmine because, lacking a charge, physostigmine is able to cross the blood-brain barrier to reverse muscarinic blockade in the CNS.
Edrophonium and Pyridostigmine.
Edrophonium [Enlon, Tensilon  ] and pyridostigmine [Mestinon] have pharmacologic effects much like those of neostigmine. One of these drugs—edrophonium—is noteworthy for its very brief duration of action. Edrophonium is also unique in that it is indicated for diagnosis, but not treatment, of MG. In current practice, though, edrophonium is not commonly used for this purpose because better and more accurate testing is now available.
] and pyridostigmine [Mestinon] have pharmacologic effects much like those of neostigmine. One of these drugs—edrophonium—is noteworthy for its very brief duration of action. Edrophonium is also unique in that it is indicated for diagnosis, but not treatment, of MG. In current practice, though, edrophonium is not commonly used for this purpose because better and more accurate testing is now available.
Drugs for Alzheimer Disease.
Three cholinesterase inhibitors—donepezil [Aricept], galantamine [Razadyne], and rivastigmine [Exelon]—are approved for management of Alzheimer disease, and one of them—rivastigmine—is also approved for dementia of Parkinson disease. With all three, benefits derive from inhibiting cholinesterase in the CNS. The pharmacology of these drugs is discussed in Chapter 18.
Irreversible Cholinesterase Inhibitors
The irreversible cholinesterase inhibitors are highly toxic. These agents are employed primarily as insecticides. During World War II, huge quantities of irreversible cholinesterase inhibitors were produced for possible use as nerve agents, but were never deployed. Today, there is concern that these agents might be employed as weapons of terrorism. The only clinical indication for the irreversible inhibitors is glaucoma.
Basic Pharmacology
Chemistry.
All irreversible cholinesterase inhibitors contain an atom of phosphorus (Fig. 12.3). Because of this phosphorus atom, the irreversible inhibitors are known as organophosphate cholinesterase inhibitors.
Almost all irreversible cholinesterase inhibitors are highly lipid soluble. As a result, these drugs are readily absorbed from all routes of administration. They can even be absorbed directly through the skin. Easy absorption, coupled with high toxicity, is what makes these drugs good insecticides—and gives them potential as agents of chemical warfare. After they are absorbed, the organophosphate inhibitors have ready access to all tissues and organs, including the CNS.
Mechanism of Action.
The irreversible cholinesterase inhibitors bind to the active center of cholinesterase, preventing the enzyme from hydrolyzing acetylcholine. Although these drugs can be split from cholinesterase, the splitting reaction takes place extremely slowly. Hence, under normal conditions, their binding to cholinesterase can be considered irreversible. Because binding is irreversible, effects persist until new molecules of cholinesterase can be synthesized.
Although we normally consider the bond between irreversible inhibitors and cholinesterase permanent, this bond can, in fact, be broken. To break the bond and reverse the inhibition of cholinesterase, we must administer pralidoxime, a cholinesterase reactivator.
Pharmacologic Effects.
The irreversible cholinesterase inhibitors produce essentially the same spectrum of effects as the reversible inhibitors. The principal difference is that responses to irreversible inhibitors last a long time, whereas responses to reversible inhibitors are brief.
Therapeutic Uses.
As mentioned previously, the irreversible cholinesterase inhibitors have only one indication: treatment of glaucoma. And for that indication, only one drug—echothiophate—is available. The limited indications for irreversible cholinesterase inhibitors should be no surprise given their potential for harm. The use of echothiophate for glaucoma is discussed in Chapter 84.
Toxicology of Cholinesterase Inhibitors
Sources of Poisoning
Poisoning by organophosphate cholinesterase inhibitors is a common occurrence. Agricultural workers have been poisoned by accidental ingestion of organophosphate insecticides and by absorption of these lipid-soluble compounds through the skin. In addition, because organophosphate insecticides are readily available to the general public, poisoning may occur accidentally or from attempted homicide or suicide. Exposure could also occur if these drugs were used as instruments of warfare or terrorism.
Symptoms
Toxic doses of irreversible cholinesterase inhibitors produce excessive muscarinic, nicotinic, and CNS effects. This condition, known as a cholinergic crisis, is characterized by excessive muscarinic stimulation and depolarizing neuromuscular blockade. Overstimulation of muscarinic receptors results in profuse secretions from salivary and bronchial glands, involuntary urination and defecation, laryngospasm, and bronchoconstriction. Prominent nicotinic effects reflect nicotinic activity at neuromuscular junctions resulting in muscle weakness, fasciculations, cramps, and twitching. Neuromuscular blockade can result in paralysis, followed by death from apnea. CNS effects may range from anxiety and confusion to delirium. Convulsions of CNS origin precede paralysis and apnea.
Treatment
Pharmacologic treatment involves giving atropine to reduce muscarinic stimulation, giving pralidoxime to reverse inhibition of cholinesterase (primarily at the NMJ), and giving a benzodiazepine such as diazepam to suppress convulsions. Respiratory depression from cholinesterase inhibitors cannot be managed with drugs. Rather, treatment consists of mechanical ventilation with oxygen.
Pralidoxime.
Pralidoxime is a specific antidote to poisoning by the irreversible (organophosphate) cholinesterase inhibitors; the drug is not effective against poisoning by reversible cholinesterase inhibitors. In poisoning by irreversible inhibitors, benefits derive from causing the inhibitor to dissociate from the active center of cholinesterase. Reversal is most effective at the NMJ. Pralidoxime is much less effective at reversing cholinesterase inhibition at muscarinic and ganglionic sites. Furthermore, because pralidoxime is a quaternary ammonium compound, it cannot cross the blood-brain barrier and therefore cannot reverse cholinesterase inhibition in the CNS. To be effective, pralidoxime must be administered soon after organophosphate poisoning has occurred.
Myasthenia Gravis
Pathophysiology
MG is a neuromuscular disorder characterized by fluctuating muscle weakness and a predisposition to rapid fatigue. Common symptoms include ptosis (drooping eyelids), difficulty swallowing, and weakness of skeletal muscles. Patients with severe MG may have difficulty breathing owing to weakness of the muscles of respiration.
Symptoms of MG result from an autoimmune process in which the patient’s immune system produces antibodies that attack nicotinicM receptors on skeletal muscle. As a result, the number of functional receptors at the NMJ is reduced by 70% to 90%, causing muscle weakness.
Treatment With Cholinesterase Inhibitors
Beneficial Effects.
Reversible cholinesterase inhibitors (e.g., neostigmine) are the mainstay of therapy. By preventing acetylcholine inactivation, anticholinesterase agents can intensify the effects of acetylcholine released from motor neurons, increasing muscle strength. Cholinesterase inhibitors do not cure MG. Rather, they only produce symptomatic relief, so patients usually need therapy lifelong.
When working with patients with MG, keep in mind that muscle strength may be insufficient to permit swallowing. Accordingly, you should assess the ability to swallow before prescribing oral medications. If the patient is unable to swallow the water, parenteral medication must be substituted for oral medication.
Side Effects.
Because cholinesterase inhibitors can inhibit acetylcholinesterase at any location, these drugs will cause acetylcholine to accumulate at muscarinic junctions as well as at NMJs. If muscarinic responses are excessive, atropine may be given to suppress them. However, atropine should not be employed routinely because the drug can mask the early signs (e.g., excessive salivation) of overdose with anticholinesterase agents.
Dosage Adjustment.
In the treatment of MG, establishing an optimal dosage for cholinesterase inhibitors can be a challenge. Dosage determination is accomplished by administering a small initial dose followed by additional small doses until an optimal level of muscle function has been achieved. Important signs of improvement include increased ease of swallowing and increased ability to raise the eyelids. You can help establish a correct dosage by having the patient or family keep records of (1) times of drug administration, (2) times at which fatigue occurs, (3) the state of muscle strength before and after drug administration, and (4) signs of excessive muscarinic stimulation.
To maintain optimal responses, patients must occasionally modify dosage themselves. To do this, they must be taught to recognize signs of undermedication (ptosis, difficulty in swallowing) and signs of overmedication (excessive salivation and other muscarinic responses). Patients may also need to modify dosage in anticipation of exertion. For example, they may find it necessary to take supplementary medication 30 to 60 minutes before activities such as eating or shopping.
Myasthenic Crisis and Cholinergic Crisis
Patients who are inadequately medicated may experience myasthenic crisis, a state characterized by extreme muscle weakness caused by insufficient acetylcholine at the NMJ. Left untreated, myasthenic crisis can result in death from paralysis of the muscles of respiration. A cholinesterase inhibitor (e.g., neostigmine) is used to relieve the crisis.
Cholinergic Crisis.
As noted previously, overdose with a cholinesterase inhibitor can produce cholinergic crisis. Like myasthenic crisis, cholinergic crisis is characterized by extreme muscle weakness or frank paralysis. In addition, cholinergic crisis is accompanied by signs of excessive muscarinic stimulation. Treatment consists of respiratory support plus atropine. The offending cholinesterase inhibitor should be withheld until muscle strength has returned.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


