Fig. 18.1
Viral vector designs
18.2.1.1 Retroviral Vectors
Retroviruses are enveloped RNA viruses of 80–130 nm in diameter with a genome size of 8–11 kilobases (kb) [11]. Retroviral genomes are made up of gag, pol, env, and 5′ and 3′ long terminal repeats (LTR). Retroviral vectors are rendered replication defective by deleting the structural, envelope, and enzymatic genes, and instead, the desired transgenes are inserted [12]. These replication-defective vectors, or proviruses, are incapable of continuing to spread after the initial infection of the target cells. The initial round of vector replication is dependent on several cis acting elements from the viral genome, including a promoter and polyadenylation signal, reverse transcription signal, transfer RNA (tRNA), primer binding site (PBS), and a polyurine tract, to initiate complementary DNA (cDNA) synthesis. Cellular tRNA binds to the PBS to initiate reverse transcription. Vectors containing the transgene are then packaged into viral particles as directed by a packaging signal (Ψ), PBS, and LTRs. Helper viruses, or plasmids, that carry the viral genes gag/pol and env, but lacking the packaging signal ψ [13], are required to express the viral proteins needed to produce vector-containing infectious viral particles. The helper function can also be provided by transfecting helper plasmids into cell cultures to produce helper cells [14]. The process of expressing the transgene in the retroviral vectors and delivering them to the target cells is called transduction [13].
Entry of the viral vectors into targeted cells is an essential first step in viral-vector-based gene delivery. Gaining entry into cells involves the interaction of the viral envelope with the host cell receptor. For lentiviral vectors, which are derived from human immunodeficiency virus type 1 (HIV1) [15], this involves a two-stage process [16]. Initially, the glycoprotein 120 (gp120) subunit of the envelope protein interacts with the target cell receptor CD4. The resulting conformational changes in gp120 allow binding with the co-receptors CXCR4 or CCR5, which belong to the chemokine family [17, 18]. Subsequently, another envelope protein subunit, gp41, signals fusion that is then followed by the release of viral capsid into the cytoplasm of the target cells. In addition to viral entry, the envelope plays an important role in determining cellular tropism by a technique called pseudotyping, where a viral vector is combined with foreign envelope proteins. Cellular tropism enhances the range of susceptible cells for the viral vectors. The lentiviral vector envelope can be substituted by the vesicular stomatitis virus G glycoprotein (VSV-G). This pseudotyping strategy has been shown not only to increase the range of possible target cell types, but also to improve stability and increase viral titers [13, 19]. Gammaretroviral vectors, e.g., murine leukemia viral vectors, transduce only dividing cells [20] and have a narrow range of cells that can have genetic material transferred to them. However, these vectors have ready access to the host genome because the nuclear membrane is removed during cell division. In contrast, lentiviral vectors transduce a wide variety of cell types, both dividing and nondividing cells such as neurons and B and T cells, albeit with varying degree of transduction efficiency [19, 21, 22].
A distinguishing feature of retroviruses is their ability to integrate into the host genome, resulting in sustained expression of the transgene. However, integration presents challenges in vector design if the vector is inserted at a site where the regulatory elements in the vector result in transcriptional activation of oncogenes [23]. Such insertional mutagenesis was the underlying cause for the adverse events in an early human gene transfer trial involving patients with severe combined immunodeficiency defect (SCID) [24]. Some patients involved in this trial subsequently developed T-cell leukemia. For lentiviral vectors, the risk of insertional mutagenesis can be reduced by designing self-inactivation (SIN) vectors that lack the viral transcriptional control elements, promoter/enhancer, and, thus, reduce the possibility of activating an oncogene located adjacent to the vector integration site [25]. Strategies have also been developed to target specific chromosomal insertion sites for the transgenes [26]. Research has also led to approaches to avoid integration. Because integration requires integrase attachment on the LTRs, mutating the integrase gene or modifying the attachment sequences of the LTRs may eliminate integration [27]. The transgene can also be expressed as episomal DNA without integration into the host genome. Such expression can occur for a relatively long duration in nondividing cells, such as retina cells [28, 29], or transiently in dividing cells.
Gammaretroviral vectors can accommodate up to 7 kb sequences [30, 31], while lentiviral vectors can accommodate larger transgenes, up to 10 kb [25]. In addition to the length of the inserted sequences, vector design must also consider stability issues. Retroviral vectors have half-lives of a few hours at 37 °C and up to a few months at −80 °C. A contributing factor to the instability is the loss of function of the envelope protein. Another factor to consider is viral titer. Lentiviral vectors can be produced at 107 transducing units/ml. The titer can be increased to 109–1010 transducing units/ml by ultracentrifugation [25]. However, titers may decrease when larger transgenes are inserted.
The first human gene transfer was conducted in 1989, as an immunotherapy to treat patients with advanced melanoma, using retroviral vectors and tumor-infiltrating lymphocytes [32, 33]. Targeted gene therapy began in 1990 with the adenosine deaminase gene for treatment of SCID [34]. Since then, numerous disorders have been treated by gene therapy [35]. They include inherited genetic disorders that involve autosomal X-linked recessive single genes or some autosomal dominant genes, and some acquired diseases, such as cancer, vascular diseases, neurodegenerative disorders, and inflammatory diseases. The first clinical trial using lentiviral vectors was approved in 2002 [36]. Currently several clinical trials are underway in which lentiviral vectors have been used to: (1) treat HIV infection, (2) transduce neuronal cells of the central nervous system for the treatment of Parkinson’s disease [37], and (3) deliver beta-globin gene for beta-thalassemia treatment [38].
There are safety concerns with the use of retroviral vectors for gene delivery. Both gammaretroviral and lentiviral vectors have the potential to illicit an immune response from the host. Immune reactions toward the viral vector result in rejection of all expressing cells. However, it should be noted that immune response is a beneficial outcome if lentiviral vectors are used to deliver genetic vaccines to treat HIV infection. Another serious safety issue is homologous recombination, which occurs when the packaging virus recombines with the vector to produce replication competent viruses. These viruses can produce harmful infections, and additionally gammaretroviruses can also cause cancer if an oncogene is activated by insertional mutagenesis [39]. In contrast, there is no evidence that lentiviral vectors result in oncogene activation through insertional mutagenesis. Another concern is the spread of the vector beyond the intended target tissue, which may cause persistent unwanted biological activity or unpredictable responses. All of these concerns have increased caution in the use of retroviral vectors although their advantages can be overwhelmingly attractive.
18.2.1.2 Adeno-Associated Viral Vectors
Adeno-associated virus (AAV) is a small, single-stranded DNA virus of 4,681 nucleotides (nt). The wild-type (wt) genome is made up of two genes, rep and cap, that encode four replication proteins and three capsid proteins, respectively. The three capsid proteins, Vp1, Vp2, and Vp3, are produced from the same open reading frame, but from differential splicing (Vp1) and alternative translational start sites (Vp2 and Vp3, respectively) [40]. Vp3 is the most abundant subunit in the virion and interacts with the host cell receptor. Recognition of Vp3 by the receptor determines cellular tropism of the virus. A phospholipase domain, essential for viral infectivity, has been identified in the unique N-terminus of Vp1 [41, 42]. The functional significance of Vp2 remains to be resolved. The viral genome is flanked on either side by 145-bp inverted terminal repeats (ITR) [43]. With the deletion of most of the viral genes, transgenes can be inserted into the cis 145-bp ITRs, to create a recombinant AAV (rAAV). The transgene can be expressed in the transduced cells without integration into the host cell genome and persists as episomal DNA [44]. The small genome size poses a limitation for AAV vector in delivering large transgenes. However, recent studies have shown delivery of genomes up to 6 kb, although delivery of these larger payloads was less efficient [43, 45]. Typically, transgenes of up to 5 kb are delivered. Strategies have been developed to improve efficiency, for example by splitting the vectors, with each vector containing approximately half of the transgene within the same cell. This approach, however, is still limited by viral packaging capacity. Additionally, cells have to be infected with different viral particles to achieve full transgene expression.
Transcapsidation is an approach to improve the packaging capacity, increase tissue tropisms, and transduction efficiency, where more than 100 different capsids from different serotypes can be exchanged to produce dozens of rAAV containing the same genome [45]. AAV serotype 2 is the best-studied AAV and was the first one used for gene transfer. However, vectors derived from alternative serotypes, e.g., 1, 4, 5, and 6 have been packaged with the same vector genome, but different viral capsids to improve efficiency and tropisms [46, 47].
rAAVs have been used in gene therapies in human muscle, liver, lung, central nervous system [48], and recently in the retina [49]. In a gene therapy trial of Parkinson’s patients, there was an improvement in the Unified Parkinson’s Disease Rating Scale (UPDRS) after 6 months for patients who received the glutamic acid decarboxylase (GAD) gene carried by an AAV2 vector that was delivered to the subthalamic nucleus [50]. rAAV2 vector carrying the gene encoding retinal pigment epithelium-specific 65-kilodalton protein (RPE65) has been used in a gene therapy trial to treat severe retinal dystrophy [51]. These results show promise, but further clinical trials are needed to demonstrate clinical significance.
18.2.2 Nonviral Delivery Systems
There has been a four-decade long history of nonviral delivery system development, prompted by on-going safety concerns associated with viral vectors. DNA transfection protocols emerged in the late 1970s, and liposome-based gene delivery strategies were first reported in the 1980s, though the field really took off in the late 1990s with the discovery of siRNA which required the use of a delivery system in vivo. Nonviral delivery systems are attractive because they typically have lower immunogenicity, lower toxicity, and their production can be easily scaled for widespread clinical use; but they also have disadvantages. These disadvantages lead to lower efficiency at each stage of the delivery process, with an overall efficiency <0.1 %.
There are a number of basic engineering considerations in the design of all synthetic vectors. They should be nontoxic, nonimmunogenic, and biodegradable, protect their nucleic acid cargo against degradation, and efficiently deliver their cargo to both the cells of interest and the desired intracellular target. Optimizing the efficiency of nonviral delivery has been the focus of much research, which includes understanding the best targeting approach for selecting the cells of interest, how the stability of formulations change after administration, and how to get efficient endosomal release. Some of the barriers to delivery are illustrated in Fig. 18.2 and include: (1) formation of nucleic acid complex, (2) entry of the complex into the cells of interest, (3) endosomal escape of the nucleic acids, (4) dissociation of the complex, and (5) transport of the nucleic acid to site of action [52]. In contrast to viral vectors, synthetic vectors are poorly optimized to take advantage of existing cellular architecture. In particular, they cannot control endosomal release, have low diffusion rate, and are unable to take advantage of active transport mechanisms. Ideally for DNA, delivery is perinuclear, and for siRNA, it is targeting of the RNA-induced silencing complex (RISC). However, the rate-limiting steps of delivery are not fully understood, and there is not a good mechanistic understanding of how to rationally optimize loading rate.
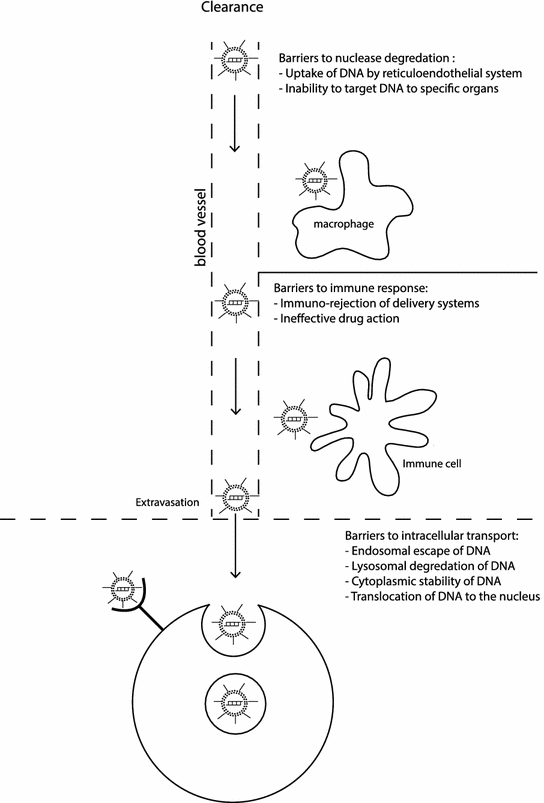
Fig. 18.2
Systemic and cellular barriers to delivery in vivo
A further challenge has been that the optimization of synthetic vectors for efficient in vitro cell culture delivery rarely translates to similar in vivo results. Monolayers of selected cells, in a carefully controlled environment, are not representative of the complex in vivo environment. The in vivo environment is a three-dimensional heterogeneous structure in an extracellular matrix, complete with enzymes, different cell morphologies, and a circulatory and immune system that cannot be easily replicated in vitro. In the worst case, some parameters can be optimized in vitro based on misleading effects which do not occur in vivo. In cell culture, size can be an advantage due to sedimentation efficiency, but in vivo smaller (less than 40 nm) particles are favored because of faster diffusion rates [53]. Another example is that positively charged vectors are beneficial in vitro for binding nucleic acids and enhancing uptake by interacting with the negatively charged cell membrane. However in vivo, negatively charged serum will bind to the vector, significantly reducing effectiveness. Some of the common designs for synthetic nanoscale delivery systems are illustrated in Fig. 18.3 and described in more detail in the following sections.
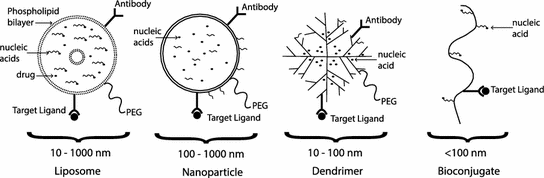
Fig. 18.3
Synthetic delivery system designs
18.2.2.1 Bioconjugation
Bioconjugation is a technique for improving delivery by covalently linking nucleic acids to bioactive targeting agents. Bioconjugation to lipids, sugars, polyethylene glycol (PEG), and peptides is in principle more attractive than delivery with cationic liposomes and cationic polymers due to the advantages of smaller-size and enhanced pharmacokinetics. They easily clear systemic circulation and, thus, can be useful for targeting oligonucleotides to cells that are not in direct contact with the vasculature such as hepatocytes. Bioconjugates of oligonucleotides have been used to deliver oligonucleotides to treat liver fibrosis [54]. Lipids, which are common targeting agents, have also been conjugated with nucleic acids for delivery of DNA, antisense oligonucleotides, and siRNA. Development of bioconjugated therapeutic drugs is more advanced; for example, site-specific anticancer drugs have been designed to consist of the hydrophobic drug linked to the nucleoside analog to form an amphiphilic bioconjugate. After cell uptake, the inactive bioconjugates or prodrugs are activated only at the target site by pH change or enzymatic cleavage.
18.2.2.2 Cell-Penetrating Peptides
Cell-penetrating peptides (CCP) are short peptides, typically arginine or lysine rich, that enhance cell uptake of an attached cargo [55]. Because of their cationic nature, they bind to the anionic glycan moieties of the extracellular matrix through electrostatic interactions [56]. Their ability to penetrate the cell membrane has been exploited to deliver large molecules, such as drugs, into cells. The positively charged CPPs are also useful for delivering negatively charged DNA or siRNA via electrostatic interactions. However, electrostatic interactions require excess CPPs, in approximately a 10:1 ratio, to maintain an overall positive charge in order to bind to glycans. A major shortcoming of CPPs as a drug delivery vehicle is their lack of specificity, but approaches have been developed to overcome this shortcoming and enhance target cell specificity. For neutral molecules, such as antisense oligomers and peptide nucleic acids (PNA), CPPs can be used as a delivery vehicle by covalent chemical conjugation. There are inherent disadvantages to using this conjugation process because the cargo may be altered as a result. Alternatively, as a way to combat this disadvantage, CPPs can form a complex with PNA, via complementary sequence hybridization, and the resulting conjugate is then used to deliver antisense oligomers to the cells.
Major families of CPPs studied in the published literature to date include: (1) penetratin, a drosophila antennapedia-derived peptide, (2) Tat peptide derived from HIV, and (3) transportan peptides [57]. These CPPs have been used to deliver therapeutic peptides that target tumor tissues by receptor-mediated endocytosis [58]. For example, the N-terminus of elastin-like polypeptides that are responsive to thermal control can be fused to CPPs. Tumor cells are specifically and selectively heated to 40–43 °C by microwave, radio-frequency, or high-intensity focused ultrasound. At these elevated temperatures, the thermally responsive elastin-like polypeptide delivery complex along with the therapeutic peptides and are taken up by tumor cells.
18.2.2.3 Liposomes
Liposomes are artificial vesicles composed of a lipid bilayer, typically in the size range of 50 nm to several microns, which self-assemble to entrap a liquid core. The surface chemistry, size, and charge of the liposome can be easily tuned through different preparation methods, and in general, they have good biocompatibility and pharmacokinetic profiles for a range of cargos. Liposomes can be fabricated with multiple concentric bilayers, with a single bilayer enclosing a liquid core, or they can form a solid or a nanostructured lipid nanoparticle [59]. Lipid nanoparticles can be fabricated down to 50 nm in size, and, as long as the formation of a crystalline structure is prevented through a blend of liquid and solid phases, there is efficient cargo loading and improved stability. However, the lipophilic core of solid particles is in general not attractive for the delivery of nucleic acids alone.
Liposomes have gained the most attention among the nonviral delivery systems because of their flexibility, biocompatibility, and tunability as well as their low toxicity, immunogenicity, and biodegradability in vivo. Two major engineering advances have helped push liposomal drug delivery products to commercial and clinical success: (1) the modification of lipids to bypass the reticular endothelial system and promote targeting, and (2) efficient cargo loading processes.
Since their first use for delivery of nucleic acids, many different types of liposomes have been developed and tested, with cationic lipids emerging as a preferred choice, often in combination with other “helper” lipids. The positively charged head groups of cationic lipids form lipoplexes with negatively charged nucleic acids, giving up to 100 % loading efficiency under the right concentrations and ratios. Endosomal escape is also primarily mediated by the cationic lipids that are believed to destabilize the endosome by forming cationic–anionic pairs with anionic lipids of the endosome membrane. This process can be enhanced by engineering the geometry of the cationic lipids and the choice of helper lipids [60].
Liposome-based delivery systems have a number of advantages, including the ability to transport large pieces of DNA, and a degree of enzyme protection in the lipoplex. Other advantages include low immunogenicity, they can be modified easily to target specific cells and their production can be scaled easily and relatively inexpensively. These advantages are offset by a number of limitations, including low overall delivery efficiency, challenges in encapsulating mixed cargoes, burst release of cargo rather than controlled release, poor storage stability, and lack of controlled release in the intracellular region of interest. For DNA delivery, ideally, this release is in the perinuclear space and the DNA can be trafficked across the nuclear envelope for efficient delivery, while for siRNA it would be close to the RISC.
Engineering of the liposome surface or the encapsulated space can address some of these limitations and increase functionality. Surface modification with PEG can provide a degree of stealth and increase circulation half-life [61], while incorporating antibodies, peptides, aptamers, affibodies, and vitamins can increase targeting efficiency [62], though it remains to be seen whether targeting robustly results in improved efficiency in humans. Mixing in helper lipids can improve fusogenicity by promoting fusion and by causing membrane destabilization [63]. Furthermore, the use of environmental triggers can increase control [64]. For example, lyso-lecithin formulations, which are thermosensitive, have progressed to clinical trials for treatment of breast and liver cancer. Photosensitive liposomes are being tested in animals and magnetosensitive liposomes are in preliminary development [59]. Environmental sensitivity can be added through enzyme-cleavable lipids and pH-sensitive lipids, both of which destabilize the liposome. Use of bioactive lipids, such as ceramide, has also been explored for tumor targeting in murine models [65]. A recent review covers many of the challenges and opportunities related to the surface chemistry of liposomes [59].
Despite nearly 50 years of research, attractive properties and many in vivo demonstrations, few liposome delivery systems have reached the market. To illustrate this point, liposomes have great potential as vehicles for DNA and peptide-based vaccines because their formulation can be adapted to protect multiple cargoes, target specific tissues, perform an immunostimulating and adjuvant role, and tuned for both humoral and cellular immune responses [66]. However, liposomes are weaker on commercially significant parameters like shelf-stability, cost-effectiveness, and the ability to scale up production reproducibly. Solid and nanostructured lipid particles that compose different lipids potentially do not share all of these weaknesses, but introduce other concerns like aggregation or coagulation.
Several engineering challenges remain for the more widespread use of liposomal delivery. Many of the studies to optimize delivery have been based on empirical observations; however, with the increasing availability of proteomic tools to understand the arrangement of transportation complexes and super-resolution microscopy to study trafficking events, a more mechanistic understanding is increasingly possible [67].
18.2.2.4 Polymers
Polymer nanoparticles can be synthetized easily in a wide range of sizes from 10 nm to several microns, and in a range of designs, including spherical and core–shell structures. Many families of polymers have been explored for nucleic acid delivery, including synthetic and natural polymers [68]. In principle, there are three ways in which polymer nanoparticles can be used for delivery: (1) by direct conjugation of the therapeutic agent with the polymer, (2) associating the agent with polymer through an electrostatic or hydrophobic interaction, or (3) by encapsulation of the agent. The most common route for nucleic acid delivery is by the formation of association complexes between the anionic backbone of the nucleic acid and a cationic polymer. The use of high buffering capacity moieties like polyanimes, so-called proton sponges, or the use of pH-sensitive components in the range pH 5–7 can also be used to aid endosomal escape to increase efficiency once captured. Furthermore, the addition of hydrophilic polymers like PEG can provide steric stability and minimize interactions in the physiological environment and reduce immune stimulation. This has driven researchers to explore heteropolymers and polymer-liposome complexes to identify efficient nonimmunogenic formations and has led to more than 35 combinations being tested so far in vivo [68].
The low molecular weight polycationic polymers, such as the polyethylene imines (PEI), are among the most commonly studied because of their small size, good association with nucleic acids, their high buffering capacity, and well-controlled chemistry. PEI/nucleic acid complexes (polyplexes) have good stability and can complex well with both siRNA and DNA, but their unmodified use is limited by stability and toxicity issues [69]. Polyarginine [70] and polylysine [71] are both lower toxicity alternatives but require additional modification for efficient in vivo use.
Naturally occurring polymers including albumin, collagen, gelatin, and chitosan have also been considered as delivery vehicles [72]. The degradation of these polymers in the presence of particular enzymes makes them attractive for controlled release. Chitosan is among the best studied and, with modification, may be attractive for oral delivery [73].
Advances in polymer chemistry have led to a number of designs incorporating functional components for enhancing efficiency and functionality, including redoxable di-sulfides, pH labile linkers, polythioketals sensitive to reactive oxygen species, and pH-sensitive hydrazone linkers [68]. The sensitivity of these smart polymers is likely to broaden over the next decade to include a wider range of physicochemical characteristics and biological processes. For controlled release of multiple therapeutic agents, the porosity of polymer nanoparticles can be tuned by formulation and manufacturing and has led to a resurgence of interest in biodegradeable polymers like the synthetic copolymer polylactide–polyglycolide (PLGA), which can be mixed with PEI for delivery of siRNA in vivo [74]. Polymeric micelles, hydrogel nanoparticles, and other protein-based nanoparticles have also been explored for drug delivery [75] and when used in combination with cationic polymers may offer some advantages for multifunctional delivery.
18.2.2.5 Dendrimers
Dendrimers are highly branched three-dimensional synthetic macromolecules, which can be produced with well-defined sizes in the range of 1–10 nm and with low dispersity. Higher generations of dendrimers resulting in the size range of 5–20 nm are favored for in vivo work because of efficient kidney filtering below 5 nm and high positive charge densities potentially creating toxicity issues and endocytosis efficiency drops as the size approaches 100 nm [76]. Their globular structure allows loading of nucleic acids and other cargos onto the outside surface via covalent bonding or electrostatic interactions to form dendriplexes, often through the use of amino groups grafted onto the end of the dendrimer branches. Poly(amidoamine) (PAMAM) dendrimers are the best studied, although they require chemical modification to reduce toxicity in vivo [77].
The small size of dendrimers makes them attractive for targeting the brain and deep tissues [76]. There have been some demonstrations of their use to deliver DNA [78] and siRNA [79] in vivo, although in vivo performance does not always match in vitro promise. By optimizing the choice of generation and surface chemistry, dendrimers do offer a potentially interesting route to cross the blood–brain barrier and deliver nucleic acid therapies to the brain.
18.2.2.6 Nanoparticles
The unique properties of materials in the nanometer to micron size range have provoked a lot of interest among physical and life scientists. For life scientists in particular, five broad classes of particles have emerged as the most studied because of their properties: (1) small particles of iron oxide, (2) quantum dots, (3) silica particles, (4) gold particles, and (5) carbon nanoparticles. The ability to design and engineer these particles to modulate their interactions with biological molecules, to carry a therapeutic cargo, and to possess characteristics favorable for imaging have led to optimism that nanometer-sized platforms can be used as multifunctional theranostics [80]. However, concerns about cytotoxicity and organ accumulation have dampened enthusiasm for the in vivo use of some of these materials.
Nucleic acids can be electrostatically immobilized on the surface of nanoparticles and protected from enzyme degradation using cationic polymers, such as the polyethylenimines or polyallylamines functionalized onto the surface of the particle. Functionalization with a mixture of polymers can also provide a degree of biocompatibility and stability in vivo. Much of the research work so far on nanoparticles has focused on in vitro and rodent studies, although to progress to human studies will require a more judicious choice of surface decoration and a better understanding of the kinetics of these particles and their nucleic acid cargoes. Some of the multifunctional imaging and delivery combinations for nanoparticles are illustrated in Table 18.1 and described in more detail in the following sections.
Table 18.1
Some of the imaging and therapeutic combinations for nanoparticle-based multifunctional delivery systems
Nanoparticle | Imaging methods | Delivery methods |
|---|---|---|
SPIONs | Optical imaging | siRNA delivery |
MRI imaging | pDNA delivery | |
PET/SPECT imaging | Drug delivery | |
Electron microscopy/X-ray imaging | Hyperthermia | |
Photoacoustic/ultrasound imaging | ||
Quantum dots | Optical imaging | siRNA delivery |
PET/SPECT imaging | pDNA delivery | |
Electron microscopy/X-ray imaging | Drug delivery | |
Photoacoustic/ultrasound imaging | ||
Silica | Optical imaging | siRNA delivery |
PET/SPECT imaging | pDNA delivery | |
Drug delivery | ||
Phototherapy | ||
Gold | Optical imaging | siRNA delivery |
PET/SPECT imaging | pDNA delivery | |
Electron microscopy/X-ray imaging | Drug delivery | |
Photoacoustic/ultrasound imaging | Hyperthermia | |
Phototherapy | ||
Carbon | Optical imaging | siRNA delivery |
PET/SPECT imaging | pDNA delivery | |
Photoacoustic/ultrasound imaging | Drug delivery | |
Hyperthermia | ||
Phototherapy |
Small Particles of Iron Oxide Nanoparticles
Small particles of iron oxide nanoparticles (SPIONs) are nanometer-sized particles of superparamagnetic magnetite and maghemite. These properties make them attractive for T2 magnetic resonance imaging (MRI), for targeting using a magnetic field and for hyperthermia. The biocompatibility and excellent in vivo imaging characteristics have led to widespread use of SPIONs for diagnostic imaging and for decorating then with different moieties, such as dyes, polymers, and peptides. Although there have been concerns about the off-target impact of these particles, a noteworthy study compared uptake of SPIONs decorated with a near-infrared dye, siRNA, and membrane translocation peptides in a mice tumor model using in vivo MRI and ex vivo optical imaging and found that there was no indication of an inflammation or cytotoxicity response [81]. Plasma DNA can also be delivered in vivo using SPIONs and imaged using MRI, fluorescence, and transmission electron microscopy (TEM) [82]. The multifunctional nature of SPIONs and their flexible surface chemistry has enabled target refinement and improvements in stability, leading to higher efficiency, as well as the delivery of a combination of anticancer drugs and siRNA in vivo [83].
Quantum Dots
At nanometer scales, semiconductor materials can possess very attractive photophysical properties, including tunable fluorescence, resistance to photobleaching, and long-term stability. The hydrophobic surface can be functionalized with polymers and decorated with peptides for targeted nucleic acid delivery in live cells [84]. Concerns about toxicity and efficiency and challenges to image deep tissues have hindered progress toward more routine use of quantum dots in vivo. In vivo work with quantum dots has focused on conjugation with antibodies for diagnostic purposes and for drug delivery [85, 86], but there may be a resurgence of interest with smaller, less toxic polymer quantum dot hybrids [87].
Silica Particles
In contrast to the other nanoparticles, silica does not offer unique imaging characteristics in the nanometer-size range, but does have excellent biocompatibility, stability and can be fabricated inexpensively into a range of shapes with different porosities, providing both interior and surface space to carry a therapeutic cargo. Some recent examples of progress in this field include a demonstration that second-generation polyamidoamine dendrimers covalently attached to the surface of 250 nm mesoporous silica particles provide protection for complexed pDNA and can successfully transfect HeLa cells in vitro [88]. Silica particles decorated with the cationic polymer polyethylenimine and loaded with siRNA have also been used to knock down EGFP expression in PANC-1 cells in vitro [89]. The porous region has been used to extend functionality beyond nucleic acid delivery to include chemotherapy agents [90], and anti-malarials [91]. The use of mesoporous silica particles to co-deliver of doxorubicin and siRNA to silence P-glycoprotein exporters in a xenograft breast cancer mouse model has recently been shown to provide synergistic tumor growth inhibition [92].
Gold Nanoparticles
The excellent chemical, physical, and optical properties of gold in the nanometer range, as well as easy synthesis of a variety of shapes and configurations has led to a long interest in gold-based synthetic vectors. Cationic polymers covalently attached to gold particles were found to be an efficient vehicle for pDNA transfection of COS-7 cells in vitro [93], and knockdown enhanced green fluorescence protein in endothelial cells in vitro using antisense oligonucleotides [94]. Increasingly, complicated designs have been developed to enhance efficiency and prolong effectiveness of siRNA using multiple layers [95], giving 70 % gene silencing of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) in a mouse model for more than 10 days post-injection [96].
Carbon Nanoparticles
Carbon has many unusual physical and chemical properties in the nanometer range and can form several different structures which are of interest as delivery vehicles. Three of these structures, nanodiamonds, carbon nanotubes (CNT), and graphene sheets have attractive features and have been explored as synthetic vectors over the past decade.
The increasing availability, innate photoluminescence, characteristic Raman signal, and good biocompatibility of nanometer-sized (~5 nm) diamonds makes them attractive for gene delivery [97–99]. In vitro delivery of siRNA into Ewing sarcoma cells has been demonstrated [99] and the attractive optical properties had led to early in vivo studies in Caenorhabditis elegans [100], although the kinetics of the functionalized nanodiamonds have not yet been fully characterized in vivo.
CNT have the ability to enter a range of cell types by passive diffusion and act as a vector [101]. A lot of effort has gone into optimizing surface modification of the CNT to enhance efficiency and biocompatibility. Hybrid cationic polymer-CNT have been demonstrated to be effective for delivering siRNA in mice at doses of <1 mg/kg with relatively high clearance [102].
Graphene sheets have high surface area and mechanical strength as well as exceptional conductivity and attractive functionalization chemistry. Oxidizing graphene followed by functionalization has emerged as an early solution to its hydrophobic nature and has been used as a siRNA vector for in vitro experiments [98]. The strong near-infrared absorption of graphene oxide has been used to demonstrate a photothermal effect in a mouse tumor model [103], and it has been used for delivery of doxorubicin in vivo [104], though more data are required to understand cellular uptake mechanisms, biodistribution, and toxicity.
18.2.3 Directed Delivery
Complementary to the carrier-mediated viral and nonviral systems described previously, delivery of nucleic acids can be enhanced locally using a range of forces, or delivered locally using a number of noninvasive and minimally invasive techniques. Local delivery or the use of fields to target regions is attractive to avoid initial systemic clearance processes, enhance cargo concentration in the region of the cells of interest, and avoid side effects associated with large doses. However, disruption of the normal physiology of the local region because of these delivery methods may have unintended consequences, including generating an inflammatory response, altering lymphatic flow and perturbation of physiological functions. Ideally, the goal for directed delivery is to combine both regional and cell-specific delivery, to minimize disturbance of neighboring cells.
18.2.3.1 Hydrodynamic Delivery
Hydrodynamic delivery, or the use of a rapid injection of fluid to deliver force in a noncompressible environment, can disrupt physical barriers, such as endothelial layers and cell membranes, to enable efficient delivery to parenchymal cells. In less than a decade, this technique progressed from first demonstration in a mouse model to use in a human clinical trial, in part because it is a simple, effective, and versatile approach and can be repeated multiple times [105]. This approach has been used for both DNA and RNA delivery to a range of tissues including liver, kidney, skeletal muscle, and myocardium, as well as tumors, through veins, arteries, and ducts. While effective in rodents, scaling hydrodynamic delivery to humans has been challenging because of concerns about the induction of irregularities in cardiac function, localized increase in blood pressure, expansion in organ size, and structural deformation, with the effects lasting up to several days in animals [105]. A number of refinements have been developed including the use of catheters, balloons, computer-controlled injectors [106], and image-guidance [107], which may address some of these challenges.
18.2.3.2 Ballistic Delivery
The ballistic delivery of nucleic acids attached to dense particles is an attractive approach for dermal targets because it is more efficient and less invasive than injection. Gene guns, using a burst of helium to give nucleic-acid-coated gold or tungsten particles enough momentum to penetrate physical barriers, emerged in the 1980s as an alternative transfection tool. For in vivo applications, the particles have enough momentum to breach the stratum corneum of the skin and have been used for vaccine delivery, but have also been used to delivery pDNA to mouse skeletal muscle cells [108]. The approach has a number of challenges, which include the risk of inflammation and damage in the target region, limited particle choices, limited nucleic acid loading capacity, indiscriminate and poor uniformity of the delivery, and sensitivity to a wide range of environmental factors. Although particle-mediated vaccine delivery systems have been tested in humans [109], it remains unclear whether cost and complexity will be barriers to wider use. However, engineering advances such as contoured shock tubes and better vaccine preservation techniques, and the lure of dose-sparing and more controlled immune response are continuing to drive interest beyond clinical studies toward regulatory approval [110]. Two other dermal delivery methods have emerged for vaccine delivery, microneedles and liquid jet injectors, both of which are reviewed in detail by Kis et al. [110].
18.2.3.3 Electric Field-Assisted Delivery
Electroporation is an extremely effective and simple technique for increasing the permeability of cells for nucleic acid delivery that has been used for more than 30 years. A number of techniques have been developed for electroporation in vivo and this technique has been used to successfully transfect liver, skin, tumor, and skeletal muscle cells [108]. Electroporation has been commonly used in DNA vaccine studies and enhanced efficacy up to 1,000-fold overnaked DNA intramuscular injections in animal models [111]. At least 10 phase I or phase II human clinical studies have been conducted with promising results [112], though with some significant adverse effects also reported that include subjects reporting pain and bleeding at the injection site [113]. At an earlier stage of development are minimally invasive electroporation devices for transdermal delivery [114] and multifunctional systems [115] that have the potential to address some of the concerns with naked DNA delivery by intramuscular injection and invasive electroporation.
18.2.3.4 Ultrasound-Assisted Delivery
Ultrasound is routinely used for in vivo imaging, although it is also known to have a therapeutic effect at higher intensities because of mechanical stimulation or disruption. Microbubbles of gas can also undergo oscillations under ultrasound stimulation disrupting the membrane and increasing the permeability of neighboring cells. This sonoporation approach has been studied for more than 25 years although all the mechanisms involved in internalization and other effects on the cell are not well used. To avoid tissue damage, relatively high frequencies (~1 MHz) and low-to-moderate mechanical indices are used, which is sufficient to cause stable cavitation of the bubbles at lower intensities and inertial cavitation at higher intensities. The 500–5,000 nm bubbles are typically composed of gas cores of a high molecular weight hydrophobic gas and a shell containing phospholipids, surfactants, targeting moieties, and nucleic acids. In vivo delivery studies have focused on organs routinely imaged by ultrasound, including successful delivery in heart, skeletal muscle, and kidney, as well as the pancreas, liver, and the central nervous system, either through direct injection into the organ or into the blood stream [116]. Both siRNA [117] and pDNA [118] have been delivered in vivo with delivery limited to the focal region of the noninvasive ultrasound. Ultrasound-mediated delivery bypasses the endocytic pathway, although there is little to no control about where the nucleic acids will enter the cell. Some concerns have been raised about efficiency [119] and that microbubbles may exacerbate underlying hypotensive reactions in some patients [120], suggesting that further optimization is required to move beyond early animal studies [121].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


