Fig. 25.1
Examples of established delivery systems. Physical techniques such as electroporation and microinjection, viral vectors in the form of retrovirus or adenovirus, as well as non-viral vectors which include liposomes, dendrimers, chitosan, and peptides are commonly used for delivery of genes/drugs into cells. Among these methods/carriers, peptides are advantageous as they are small in size, easily synthesized and modified, can condense DNA into compact particles for transport, preserve bioactivity of the drug until it is delivered to the target site, disrupt the endosomal membrane and escape proteasomal degradation, traffic therapeutic molecules of various size, charge and function to targeted intracellular compartments, and additionally, have reduced cytotoxicity and immunogenicity
Aside from the useful qualities exhibited by peptides, the ability to deliver bioactive molecules and drugs in a slow, sustained, controlled release format is also important. The applicability of such a delivery system would be further expanded if the carriers were also biodegradable, biocompatible, mechanically durable, and could be prepared and processed under ambient aqueous conditions to preserve the bioactivity of the drug to be delivered. Silk proteins are promising biomaterials that can fulfill these needs. In addition, they have advantages of slow biodegradability, biocompatibility, self-assembly, excellent mechanical properties (tensile strength and Young’s modulus), and controllable structure and morphology [17, 18]. Silk proteins have been produced by spiders and insects such as silkworms, and they form fibrous materials. Recombinant silks are also synthesized by the elucidation of silk genetics, structures, and biophysics.
We describe in this chapter peptide/protein-based carriers for intracellular gene or drug delivery as well as organelle-targeted delivery and highlight some of the recent advances in designs. Diverse applications where these peptides have been harnessed are presented, as well as how these carriers have helped to advance the field of cell biology. The use of silk-based biomaterials to deliver bioactive molecules, such as small drugs, proteins, and genes, is also described in this review.
25.2 Peptide-based Drug/Gene Carrier
Peptides possess many advantages as delivery vehicles in comparison with viral vectors and outperform even other non-viral vectors with the small size, ease of synthesis and modification, and good biocompatibility. A highly versatile and efficient class of transporters, known as cell-penetrating peptides (CPPs), comprises short peptide sequences (6–30 amino acids) with the ability to traverse plasma membranes. CPPs vary significantly in sequence, hydrophobicity, and polarity. These peptides are typically cationic and exist as naturally occurring protein transduction domains or can be rationally designed [14] (Table 25.1). Tat [19] represents CPPs derived from naturally occurring proteins. A second group contains chimeric CPPs such as transportan, which has 12 amino acids derived from the neuropeptide galanin fused with 14 amino acids derived from the wasp venom mastoparan [20]. A third group contains synthetic CPPs, and of these the polyarginines [21] are the most studied. Identification of the first CPP, the trans-activating transcriptional activator (Tat) of HIV-1, dates back to 1988 from two research groups working on HIV [22, 23]. Other CPPs have been discovered since, and the list continues to grow.
Table 25.1
Examples of cell-penetrating peptides described in this review, sequences and origins
Peptide | Sequence | Origin | Reference |
|---|---|---|---|
Tat (48–59) | GRKKRRQRRRAP | Protein-derived | [19] |
Penetratin | RQIKIWFQNRRMKWKK-NH2 | Protein-derived | [42] |
pVEC | LLIILRRRIRKQAHAHSK-NH2 | Protein-derived | [72] |
PTD4 | YARAAARQARA | Protein-derived | [82] |
Transportan | GWTLNSAGYLLGKINLKALAALAKKIL-NH2 | Chimeric | [20] |
PepFect6 | Stearyl-AGYLLGK(εNHa)INLKALAALAKKIL-NH2 | Chimeric | [37] |
Tetravalent decaarginine (10R-p35tet) | R10GEYFTLQIRGRERFEMFRELNEALELKDAQA | Chimeric | [57] |
Bombesin (6–14) | NQWAVGHLM-NH2 | Chimeric | [109] |
Pep-1 | Acb-KETWWETWWTEWSQPK KKRKV-cyac | Synthetic | |
Polyarginine | R n (n = 6–12) | Synthetic | [21] |
CADY | Ac-GLWRALWRLLRSLWRLL WRA-cya | Synthetic | [40] |
YTA2 | YTAIAWVKAFIRKLRK-NH2 | Synthetic | [73] |
YTA4 | IAWVKAFIRKLRKGPLG-NH2 | Synthetic | [73] |
Although the mechanism underlying the internalization of CPPs across the cellular membrane remains to be elucidated, recent studies have helped to shed some light on the subject [24]. To date, these peptides have successfully delivered a wide range of exogenous molecules including nucleic acids, proteins, antibodies, nanoparticles, and liposomes (Fig. 25.2). An intense and ongoing subject of research is the intracellular delivery of genes due to their tremendous potential as therapeutic agents for many acquired or inherited diseases. The selective permeability of the plasma membrane presents a major challenge for internalization of these large and negatively charged molecules. Unassisted uptake of nucleic acids, if at all, occurs only at negligible levels [25]. Cationic CPPs, on the other hand, are not only able to destabilize the cell membrane but can also bind and compact nucleic acid molecules via electrostatic interactions [26], thereby aiding the translocation of these molecules across the cell membrane.
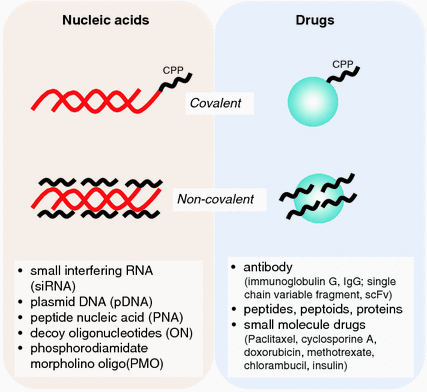
Fig. 25.2
Covalent and non-covalent linkages of cell-penetrating peptide (CPP) to various drug and nucleic acid cargoes
For efficient uptake, the size of CPP–nucleic acid complexes should preferentially be in the range of 100–300 nm. An equally important criterion is the stability of complexes which can be regulated by varying the charge ratio, as overly strong electrostatic interactions may prevent dissociation of nucleic acids from the carrier inside the cell [27]. Although CPP–nucleic acid complexes face the risk of being trapped in endosomes, the current state of research on CPPs enables modifications of peptides to improve their endosomal escape [28–30]. Most importantly, CPPs have been found to have minimal cytotoxicity at concentrations required to mediate the delivery of various cargoes [31]. Additionally, the versatility of CPPs lies in their ability to undergo structural modifications to suit different types of cargoes. For instance, CPPs can be conjugated covalently as well as non-covalently with siRNAs [32, 33].
The discovery of siRNA as a powerful strategy for the modulation of gene expression [34] led to various approaches to modify the systems to optimize potency, specificity, and delivery [35, 36]. The past few years have seen the emergence of new CPPs such as PepFect6 (variant of TP-10) [37], gastrin-related peptides [38], and bLFcin6 (derived from bovine lactoferricin) [39], specifically designed for optimized delivery of siRNA. The first secondary amphiphilic peptide, CADY, showed improvement in terms of higher affinity for siRNA and higher viability (more than 80 %) with all tested cell lines [40]. Tat peptide, crosslinked to siRNA, effectively increased cellular uptake of the oligonucleotide [41]. The degree of silencing achieved by Tat-mediated siRNA delivery matched that using the highly efficient transfection reagent, Lipofectamine 2000. Reduced gene expression for up to 7 days could be attained with penetratin [42] and transportan covalently linked to modified siRNA [43], while CPPs in the form of polyarginine and an MPG derivative formed non-covalent complexes with siRNA for delivery [44, 45]. A separate study boasted an efficient and persistent silencing effect with minimum cytotoxicity using siRNA packaged in octaarginine-modified MEND (R8-MEND) for cell transfection [46]. In another report, a type of CPP termed “peptide for ocular delivery” (POD) successfully transported siRNA into human embryonic retinal cells to result in a gene silencing effect of more than 50 % [47]. POD also delivered plasmid DNA (pDNA) to cells with gene expression exceeding 50 %. In addition, macrobranched Tat [48], arginine- or histidine-rich CPPs [49, 50] and some well-known synthetic peptide analogs such as GALA [51], KALA [52], JTS1 [53], and ppTG1 [54] were found to function efficiently as carriers of pDNA. Multimers of Tat increased transfection efficiency of plasmids by 6–8 times more than poly-l-arginine or mutant Tat2-M1 and by 390 times compared with other standard vectors [55]. We recently developed peptide carriers, which were fusions of different CPPs and polycations, as a new tool for gene delivery (Fig. 25.3). The designed peptides facilitated rapid delivery of pDNA and showed better transfection efficiency compared to either just the CPP or polycationic region alone [56]. Related to the idea of a fusion peptide, multivalency also improves the activity of CPP. Tetrameric CPPs performed 10–100 times better in cellular uptake and mediated as high as a 1,000-fold increase in activity relative to their monomeric counterparts [57].
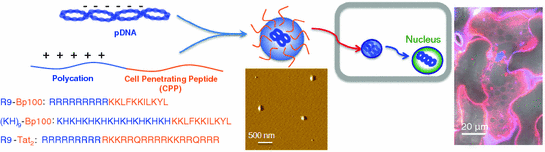
Fig. 25.3
A useful strategy for rapid and efficient gene delivery which is based on the combination of a cell-penetrating peptide (Bp100 or Tat2) and a polycation (nona-arginine or a copolymer of histidine and lysine) (adapted from [57])
Apart from siRNA and plasmids, CPPs have also been used widely in the delivery of DNA mimics such as peptidic nucleic acid (PNA), decoy oligonucleotide (ON), and phosphorodiamidate morphilino oligomer (PMO). CPP (transportan or penetratin) conjugates of PNA analog exhibited inhibition of Tat-dependent trans-activation in HeLa cells within 6 h when coadministered with chloroquine [58]. Nuclear localization of splice-correcting ON analogs was accomplished by CPP-mediated delivery [59]. A specific example is the delivery of double-stranded decoy ONs targeting the transcription factor nuclear factor kappa-light-chain-enhancer of activated B cells and Myc protein by TP or TP10 [60, 61]. R8-MEND, which can transport siRNA, also transported antisense oligodeoxynucleotides to functionally inhibit cytosolic mRNA [62]. CPPs have also been applied for PMO delivery into mouse models of Duchenne muscular dystrophy, to enable splice correction of a nonsense mutation at exon 23 in the dystrophin gene [63, 64].
Intriguingly, the Rath peptide, derived from VP5 protein of avian infectious bursal disease virus, rapidly delivered not just DNA but also antibody within 30 min for the former, and 1 h for the latter [65]. Similar to their application in nucleic acid delivery, CPPs are just as commonly used for cellular delivery of a variety of drugs, with sizes ranging from 30 to 150 kDa. CPPs can deliver these molecules by chemical modifications, non-covalent transduction, or using fusion proteins. Polyarginine conjugates of paclitaxel enhanced the water solubility, and thus cellular uptake of this hydrophobic drug [66, 67]. The same CPP, when conjugated with cyclosporine A, showed potential use for treatment of psoriasis as well as other skin diseases [21]. Penetratin, on the other hand, made delivery of photoactive drugs possible by mitigating their extreme lipophilicity and poor trafficking properties [68].
Some of these CPPs also play a pivotal role in cancer therapy. Doxorubicin conjugated to Tat and transferrin successfully inhibited the growth of multidrug-resistant tumor cells [69, 70]. This well-characterized chemotherapeutic was also employed to evaluate the anti-tumor efficacy of a new and potential glioma-targeted drug delivery vector, gHoPe2 [71]. In this study, the authors reported construction of the new vector by covalent conjugation of pVEC [72], gHo, and a cargo, and the efficiency of drug delivery by gHoPe2 highlighted the feasibility of the strategy. In another study, two different CPPs, YTA2 and YTA4, increased uptake of methotrexate in resistant tumor cells [73]. CPPs are invaluable tools in cancer therapy primarily because they can be modified to improve selectivity for cancer cells. Conjugation of a Tat derivative to AHNP, a mimetic that binds selectively to an epidermal growth factor receptor overexpressed in breast cancers, yielded a cell-specific transporter [74]. Subsequently, this transporter successfully delivered the intended therapeutic to cancer cells both in vivo and in vitro, without compromising the activity of the drug. Tumor-homing domains can also be engineered into CPP, for instance, a PEGA-homing domain known to accumulate in breast tumor vasculature was conjugated to pVEC CPP, and the resultant construct was effective and selectively taken up into breast tumor blood vessels [75]. Conjugation of a chemotherapeutic agent, chlorambucil, to the same CPP enhanced efficacy of the drug as much as 4 times.
In comparison with the delivery of small molecule therapeutics for the treatment of cancer and other diseases, a more challenging task is the translocation of large molecules such as antibodies into cells. Tat peptide, known for its remarkable translocation capacity, was first exploited for such a function two decades ago [76]. Since then, Tat has been used to deliver 111In-labeled anti-mouse IgG for radiotherapeutic applications [77] and anti-p21 for sensitizing cancer cells to cytotoxic therapies [78]. Some recent achievements include the delivery of single-chain antibody fragments into mouse brain cells to target prion diseases [79] and into hepatocytes to inhibit hepatitis B virus replication [80]. The ability of CPPs to overcome various barriers is showcased with examples, such as the blood–brain barrier, membrane barrier of hepatocytes and, additionally, the intestinal barrier for the delivery of insulin [81]. An impressive point to note was that covalent attachment of the CPP to insulin was not required; instead, the simple coadministration sufficed. Bioactive macromolecules in the form of peptides, peptoids, and proteins have also been successfully delivered to modulate intracellular processes. A representative example for each of these different types of cargo is described. The peptide inhibitor of transcription factor STAT-6 was conjugated to the HIV-Tat-derived PTD4 CPP [82], and internalization of the conjugate attenuated ovalbumin-induced inflammatory responses and mucus production in mice [83]. Anti-apoptotic proteins, Bcl-xL and its BH4 domain, were conjugated to Tat for administration to mice to treat bacterial sepsis, which pioneered the use CPP in such applications [84]. The conjugation of Tat and Penetratin, individually, to a peptoid inhibitor improves cell membrane permeability, with higher inhibition observed using the penetratin conjugate [85]. Antigens, with ovalbumin as a model, were conjugated to the translocation motif of the hepatitis B virus for intracellular delivery [86]. The authors observed improvements of both cellular and humoral immune system responses. Interestingly, it is possible for a single carrier, such as the amphiphilic peptide Pep-1, to promote cellular uptake of both small peptides as well as large proteins regardless of their nature and target cell type [15, 87].
25.3 Organelle-Specific Delivery System
The ultimate goal in designing a peptide carrier is to make one with target specificity or the ability to carry therapeutics not just to a specific cell, but to a specific organelle within the cell. This is crucial as a majority of therapeutic agents need to be in particular intracellular compartments to fully exert their therapeutic potential. Some diseases and corresponding treatments have been associated with specific organelles—the cell nucleus is a well-known target for gene therapy; the mitochondrion itself is important for proapoptotic cancer therapy, while its genome is a prime target for manipulation; and the lysosomes are recognized for targeting of enzymes to treat lysosomal storage diseases. An effective strategy for organelle-specific targeting is by the use of signal peptides, which have specific amino acid sequences for recognition by the cellular machinery in order to direct attached cargoes toward correct destinations in the cell [88] (illustrated in Fig. 25.4).
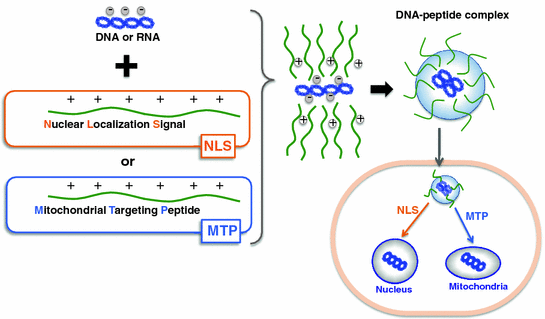
Fig. 25.4
Peptide carrier loading and targeting strategies for gene delivery. Non-covalent complex of signal peptides (nuclear localization signal or mitochondrial targeting peptide) and cargo (DNA or RNA) for localization to specific organelles (nucleus or mitochondria). The complex is formed by electrostatic and hydrophobic interactions between the peptide and the cargo
The most established system for transport of molecules between the cytoplasm and the nucleus is the classical nuclear import pathway, whereby proteins containing nuclear localization signals (NLS) are transported into the nucleus. Since characterization of the NLS from simian virus 40 (SV40) large T antigen in 1984 [10], this and other NLS peptides have been used to traffic DNA to the nucleus [89]. These NLS peptides ensure entry of DNA through the nuclear pore complexes by active transport as free diffusion is an unlikely route for DNA passage due to size restrictions [90, 91]. NLSs are characterized by short clusters of basic amino acids and are typically about 10 amino acids in length. They form complexes with DNA by ionic interactions, covalent attachments, or site-specific attachment using a peptide nucleic acid clamp [92]. In the field of gene therapy, the most notable NLS originated from the large tumor antigen of SV40. This peptide of 7 amino acids enabled a 100-fold increase in the speed of nuclear uptake of pDNA compared to a reversed NLS sequence [93]. In another study, the M9 sequence of the heterogeneous nuclear ribonucleoprotein attached to DNA in a non-covalent manner improved nuclear import of DNA. Conjugation of the M9 sequence to a cationic peptide for DNA condensation resulted in a 10-fold increase in reporter gene expression [94]. As nuclear-targeted drug delivery can kill cancer cells more directly and efficiently, some recent work featured design strategies using different NLSs and doxorubicin as model drug. In one of the studies, Tat was conjugated with mesoporous silica nanoparticles and was found to efficiently target the cell nucleus, killing cancer cells effectively [95]. Another study reported on a novel nucleus-targeting carrier comprising NLS conjugated to cholesterol-modified glycol chitosan, which facilitated nuclear localization of higher doxorubicin amounts than unassisted uptake of the drug [96].
As mentioned earlier, mitochondria represent another important target for intracellularly delivered DNA and drugs. Not only is this organelle responsible for ATP production, the mitochondrion is also involved in various metabolic pathways essential for the regulation of fundamental cellular processes. Mitochondria dysfunction therefore contributes to a variety of disorders and diseases namely cancer [97], Parkinson’s disease [98], Alzheimer’s disease [99], diabetes [100], and atherosclerosis [101], among others. Since the discovery of peptides with mitochondria-targeting ability, a variety of innovative approaches to develop new peptides or fine-tune properties of existing peptides to achieve efficient mitochondrial localization have surfaced. Introduction of lipophilic residues and modification of charge distribution in a CPP yielded peptides with mitochondrial targeting ability [102]. A different approach was taken in a separate study where artificial signal sequences were created, and the fabricated sequences worked favorably to target the mitochondria just as well as natural signal sequences [12]. Zhao and coworkers successfully used tetrapeptide sequences containing radical scavenging properties to deliver antioxidants to the mitochondria. The versatile Tat peptide, when fused to a mitochondrial targeting sequence from malate dehydrogenase, efficiently translocated proteins across mitochondrial membranes [103]. These mitochondrial targeting peptides (MTPs) are typically N-terminal motifs predicted to form amphipathic helixes approximately 15–70 residues in length and enriched in positively charged basic residues [104]. To date, different MTPs have been discovered for the delivery of various cargoes. The DNA alkylating drug chlorambucil was delivered as an MTP conjugate [105], while histatin 5, an anti-microbial peptide, can be used as a carrier molecule for leishmanicidal molecules to target mitochondria ATP synthesis in Leishmania [106]. Fusion of adeno-associated virus capsid with a mitochondrial targeting sequence provided a platform for introduction of large DNA fragments (~5 kb) into mitochondria [107], which was applied in a follow-up study for mutant gene delivery to successfully induce visual loss and optic atrophy in mice [108]. In other new cancer-related studies, a chimeric peptide analog of bombesin targeted mitochondria-disrupting peptides to the mitochondria in tumor cells [109], while a combination of mitochondria-targeting genes (LETM1 and CTMP) and 2A-peptide sequence induced anti-tumor effect and mediated mitochondria apoptosis [110].
In the case of lysosomes, delivery of enzymes is the only means of treatment for lysosomal storage diseases (LSDs) such as glucocerebrosidase, phenylalanine ammonia lyase, and many others. More than forty of the known LSDs are caused, directly or indirectly, by the absence of one or more enzymes in the lysosome [111]. Hence, enzyme replacement therapy for LSDs is being actively pursued although advancement in the area still falls short compared to well-established therapeutic strategies for diseases associated with the mitochondria or nucleus. Nevertheless, recent progress related to lysosomal targeting is highlighted. LeBowitz and coworkers tested an alternative, peptide-based targeting system for delivery of enzymes to lysosomes. Their strategy involved a chimeric protein containing a portion of mature human IGF-II fused to the C terminus of human β-glucuronidase, which was delivered effectively and reversed the storage pathology in mice as models [11]. The same research group reported recently on lysosomal targeting of acid α-glucosidase to enhance muscle glycogen clearance in mice with Pompe disease, where human clinical studies are currently underway [112]. Other researchers evaluated subcellular targeting and delivery of nanoparticles to the lysosome in mammalian cells using a novel carrier comprising cell-penetrating peptide and lysosomal sorting peptide domains [113]. Another approach taken in the development of new vectors was to synthesize, by click chemistry, an inhibitor–Tat conjugate, which was successfully delivered to lysosomes for inhibition of lysosomal cysteine proteases [114].
25.4 Drug Delivery Via Silk Nanoparticles
Silk proteins have been used successfully for several decades as sutures and have also been explored as biomaterials for drug delivery systems, due to their excellent mechanical properties, versatility in processing, and low cytotoxicity [18, 115, 116]. The degradation products of silk proteins with beta-sheet structures, when exposed to alpha-chymotrypsin, have been identified and shown to have no cytotoxicity to in vitro neuron cells [117, 118]. The combination of silk protein and nanoparticles makes useful delivery systems because silk nanoparticles are advantageous over other delivery methods in terms of biodegradability and biocompatibility. In this section, silk-based nanoparticles and their preparation methods, which have been widely studied by many groups, are summarized.
Silk nanoparticles from silk fibroin solutions of Bombyx mori and tropical tasar silkworm Antheraea mylitta were stable, spherical, negatively charged, 150–170 nm in average diameter, and showed no toxicity [119]. The silk nanoparticles were observed in the cytosol of murine squamous cell carcinoma cells, and the growth factor release from the nanoparticles showed significantly sustained release over 3 weeks, implying potential application as a growth factor delivery system [119]. Silk nanoparticles were also used to increase mechanical properties of silk-based porous scaffolds [120]. Several types of silk nanoparticles hybridized with chitosan, sericin, elastin, and genes have been developed due to their excellent availability and biocompatibility [121–125]. Silk fibroin-based nanoparticles (<100 nm) for local and sustained therapeutic curcumin delivery to cancer cells were fabricated by blending with non-covalent interactions to encapsulate curcumin in various proportions with pure silk fibroin or silk fibroin with chitosan. The silk-based nanoparticles containing curcumin showed a higher efficiency against breast cancer cells and have potential to treat in vivo breast tumors by local, sustained, and long-term therapeutic delivery [126]. Silk sericin–poloxamer nanoparticles loaded with both hydrophilic and hydrophobic drugs were reported to be stable in aqueous solution, small in size (100–110 nm), and rapidly taken up by cells [122]. Conjugation of bioactive agents such as drugs or peptides into silk-based nanoparticles is also an efficient methodology for delivery of these molecules into target sites. The crystalline silk protein nanoparticles (40–120 nm) have been conjugated with insulin via covalent crosslinking [127]. It was confirmed that in vitro stability of insulin in human serum was enhanced and the half-life of insulin prolonged by conjugating with silk protein nanoparticles. Silk fibroin was also bioconjugated with L-asparaginase to form crystalline nanoparticles with diameters of 50–120 nm [124]. These bioconjugates had an increased resistance to trypsin digestion, better stability in serum, storage stability in solution, and no leakage of the enzyme from the nanoparticles.
The reconstituted dragline silk proteins from the spider Araneus diadematus have been used to prepare microcapsules for drug delivery using self-assembly of the proteins at an emulsion interface [128]. These microcapsules were suggested to be useful to encapsulate small active ingredients, provided that the active ingredient does not impede the adsorption of the silk and/or that the encapsulation process does not alter the ingredient [128]. Microspheres of bioengineered spider silks, which were derived from ADF4 from A. diadematus, were formed by several methods such as dialysis and micromixing [129]. As a result of their material strength, biocompatibility, and the possibility of functionalization via recombinant protein techniques, spider silk microspheres may offer potential for the development of targeted drug delivery systems.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


