Fig. 11.1
Structure of an antibody (IgG1). IgG1 consists of two heavy chains (gray) and two light chains (black) which are connected by disulfide bonds. The heavy chain contains three constant domains (CH1–CH3), a variable domain (VH), and a hinge region between CH1 and CH2. The light chain contains a constant (CL) and a variable (VL) domain
Most therapeutic mAbs are of the γ-immunoglobulin (IgG) isotype. IgG has a particularly long half-life in serum, due to a recycling mechanism based on the interaction with the neonatal Fc receptor (FcRn). Endocytosed IgG binds with its Fc part to the FcRn at pH 6.0–6.5 in the endosomes and recycles to the blood serum. IgG contain a single N-glycosylation site in the constant region of each heavy chain. The attached glycans have been shown to play a crucial role for some of the effector functions, but also contribute to the stability of the Ab. There are four different subtypes of IgG in humans (IgG1–IgG4), which have different structural features and biological properties.
11.2 Reducing Immunogenicity of Therapeutic Antibodies
Rodent Abs The in vitro production of murine mAbs from hybridoma cell lines first made it possible to use this class of molecules as therapeutics [7]. However, murine mAbs (suffix: -omab) are immunogenic in humans and induce human anti-mouse Abs (HAMA) [8]. This precludes repeated administration of murine mAbs and therefore long-term therapy. In addition, the half-life of murine mAbs in humans is relatively short, and they are not efficient in recruiting effector functions. Despite these difficulties, several murine mAbs have been approved by the FDA, primarily for diagnostic use.
Chimeric Abs A first attempt to reduce the immunogenicity of murine mAbs was the design of chimeric mouse–human Abs (suffix: -ximab). The variable regions of rodent heavy and light chains were grafted onto the constant regions of human IgG1 by recombinant DNA techniques [9–11]. Chimeric mAbs exhibited a longer half-life in humans and were less immunogenic than murine mAbs. In addition, the isotype relevant to the desired biological function of the mAb, typically IgG1, could be chosen by the chimerization technique. Several chimeric mAbs have been approved as therapeutics, for example, cetuximab, rituximab, or infliximab. However, about 33 % of the sequence is still of mouse origin and induce the human anti-chimeric antibody (HACA) response [12].
Humanized Abs To confine the potential immunogeneicity of therapeutic mAbs, further reduction of the mouse sequences was approached by various methods. Humanized Abs (suffix: -zumab) obtained by CDR–grafting contain only the hypervariable regions from the original mouse mAb [13]. This method often requires the introduction of some amino acids from the original mouse sequence into the framework regions of the variable domains, in order to retain the binding properties of the original Ab in a process termed reshaping [14]. CDR grafting results in humanized Abs with about 95 % of the sequence of human origin (Fig. 11.2).
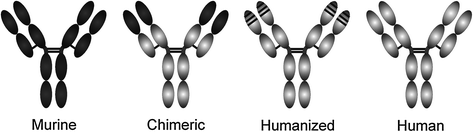
Fig. 11.2
Monoclonal antibody types. Murine sequences are shown in dark, human sequences in light gray
Further reduction of the murine sequences can be achieved by SDR transfer instead of CDR grafting. Only 20–33 % of the CDR residues have been found to participate in antigen binding. These “specificity determining residues” (SDR) were successfully grafted to human Ab sequences with “abbreviated CDRs” [15]. Alternatively, in a process termed veneering or resurfacing, only the surface residues of the framework regions of murine variable domains are replaced with human residues [16, 17], while the remaining sequences, including CDRs and buried amino acids, remain murine.
DeImmunization Deimmunization is a technique by which potential linear T cell epitopes in Ab sequences are predicted by bioinformatic approaches [18]. Specifically, known human HLA class I or class II epitopes are identified in silico. Potential T cell epitopes can then be altered by appropriate amino acid substitution. A similar approach aims at the removal of both T and B cell epitopes using human string content (HSC) [19].
Human Abs Fully human Abs (suffix: -umab) can be generated by in vitro display techniques, including phage display [20–22] or yeast display [23]. Phage display libraries consist of a large diversity (>109) of filamentous bacteriophage, each expressing a different Ab fragment. Typically, Fab fragments or single-chain variable fragments (scFv) are presented on the phage. The diversity of such in vitro libraries can either be derived from a human antibody repertoire or generated synthetically. The library is screened against the target antigen to enrich for target-binding Ab fragments. This procedure usually requires several rounds of selection to obtain high-affinity Ab fragments. In vitro selection of Abs allows close control over stringency and specificity. For example, it is possible to include steps into the selection strategy that allow enrichment of Abs that cross-react with the rodent variant of the antigen or to deselect Abs that cross-react with homologous proteins.
Alternatively, fully human mAbs can be generated in transgenic mouse strains with inactivated mouse immunoglobulin genes which have been replaced with the corresponding human gene sequences [24–27]. Many different transgenic mouse platforms have been developed and yielded human mAbs for clinical development. Today, human mAbs are the fastest growing category of mAb therapeutics entering clinical studies [28].
11.3 Engineering Pharmacokinetics
IgG have a relatively long half-life in blood, ranging from 7 to more than 16 days. One major goal of Ab engineering is to further prolong the half-life of therapeutic mAbs in circulation, in order to maintain therapeutic mAb levels and to allow for less frequent dosing. On the other hand, a shorter half-life may be of interest for radiolabeled Abs, for example, to control toxicity or to reduce background radiation in imaging. Mutations have been introduced in the Fc part of IgG1 in order to increase binding to FcRn under acidic conditions. These mutations lead to an optimized recycling of the engineered mAb through FcRn binding in endosomes and thus to a longer half-life [29, 30]. Further, the fast-clearing istoypes IgG2, IgG3, and IgG4 have been engineered in their Fc part to improve half-life and stability in blood [31–33].
A variable that contributes to the non-specific clearance of mAbs is their isoelectric point (pI). Non-specific clearance is a result of pinocytosis of Ab into the endosomes and subsequent lysosomal degradation of the non-FcRn-bound Ab [34]. A higher pI generally results in a faster clearance of the mAb [35]. Lowering the pI by engineering the variable region is therefore a promising approach to increase the half-life of some mAbs [36]. In addition to mutagenesis, chemical modification has been used to improve the pharmacokinetics by altering the pI [37].
Finally, antigen-mediated clearance may contribute to the clearance rate of an mAb. Similar to the FcRn binding described above, Abs have been engineered for pH-dependent antigen binding [38]. These Abs maintain high binding affinity at neutral pH, but dissociate rapidly from their target in the acidic environment of the endosomes. The antibody then recycles back to the cell surface and is rescued from lysosomal degradation [39, 40].
11.4 Enhancing Therapeutic Efficacy
Monoclonal antibodies are an important class of cancer therapeutics and widely used in the clinics today. However, they often are not sufficiently effective as monotherapies and need to be combined with chemotherapeutics. Hence, Abs have been engineered to become more potent cytotoxic drugs. There are several strategies that aim at the generation of more cytotoxic Ab constructs. These include engineering to enhance intrinsic effector functions or the introduction of an additional cytotoxic moiety attached to the Ab. In the latter cases, the Ab’s effector functions play a subordinate role as does the biological significance of the target for disease development. The Ab simply becomes a vehicle which delivers cytotoxic payload to the tumor cells.
11.4.1 Fc Engineering to Enhance Effector Functions
IgG are able to induce ADCC by interaction with the FcγR which is expressed on cytotoxic T lymphocytes and natural killer cells. Improving the binding of the IgG to FcγR could therefore improve the therapeutic efficacy of mAbs where these effector functions are the mechanism of action to destroy the target cells.
The Asn297-linked glycans of an IgG play an important role in the interaction with the FcγR. It has been demonstrated that engineering of the glycan structure by the presence of a bisecting N-acetylglucosamine or the absence of fucose enhances the binding to the FcγRIII and consequently improves ADCC [41, 42]. The glycoengineered Abs are obtained from mutant CHO cell lines which are expressing or lacking the corresponding sugar-modifying enzymes.
11.4.2 Antibody–Drug Conjugates
Antibody–drug conjugates (ADCs) combine the specificity and targeting properties of an mAb with the potency of a cytotoxic drug [45]. Typically, a cytotoxic small molecule is chemically linked to an mAb which binds to an antigen expressed on the cell membrane of cancer cells. Upon binding, the ADC–antigen complex is internalized into the cell and transported along the endosomal–lysosomal pathway. The attached drug is eventually released from the ADC and becomes active within the cell by interfering with vital cellular processes. It is a prerequisite that the cytotoxic drug is extremely potent and exhibits its cytotoxic potential at low concentrations. While early ADCs employed classical chemotherapeutics as drugs such as vinblastine, doxorubicin, or methotrexate, typical drugs now include highly potent microtubule-targeting agents (auristatins, maytansinoids) or DNA-damaging drugs (calicheamicins, duocarmycins). Due to their high systemic toxicity and lack of specificity for cancer cells, these drugs cannot be used as single-agent therapeutics.
To ensure controlled release of the drug within the target cells, a linker has to be introduced between the mAb and the cytotoxic drug [46]. These linkers have to be stable in circulation to prevent spontaneous release of the drug and consequential damage to non-targeted tissue. Current linker strategies include the following:
Acid-labile hydrazone linkers, which are cleaved in the low-pH environment of the endosomes (pH 5.0–6.5) and lysosomes (pH 4.5–5.0), but relatively stable in the bloodstream (pH 7.3–7.5).
Disulfide-based linkers, which are cleaved in the reducing environment of the cytosol.
Non-cleavable thioether linkers, which depend on the degradation of the mAb in the lysosome and result in the release of drug still attached to the linker and the amino acid (usually lysine) to which the drug has been attached [47].
Dipeptide and thioether linkers are more stable in circulation than the chemically cleavable hydrazone or disulfide-based linkers. However, the cytotoxic metabolite resulting from the cleavage of thioether-linked ADCs is charged and has been shown to be less cytotoxic on neighboring cells than the corresponding product from a cleavable linker.
Wyeth’s Mylotarg (anti-CD33 humanized IgG4 mAb conjugated to a calicheamicin by a hydrazone linker) was the first ADC to reach the market [50]. It was withdrawn by Pfizer in 2010 because of limited clinical benefits and safety concerns. Adectris (Brentuximab Vedotin) and anti-CD30 chimeric mAb conjugated to auristatin E (MMAE) received FDA approval in 2011 [51] for the treatment of Hodgkin lymphoma and anaplastic large-cell lymphoma. Recently, FDA approved the maytansinoid-based ADC Trastuzumab emtansine (T-DM1, Roche/Genentech) for breast cancer [52]. The three ADCs that have reached the marked so far are different in linker strategy, target antigen, and drug (Table 11.1). Many ADCs are now in early-stage clinical testing [45]. Future developments address new highly potent drugs, more uniform production of ADCs, and the better penetration of solid tumors by ADCs, for example, by using antibody fragments or non-IgG scaffolds.
Table 11.1
Antibody–drug conjugates that reached the market by 2013
ADC | Status | Antigen | Linker | Drug | |
|---|---|---|---|---|---|
Mylotarg | Gemtuzumab ozogamicin | Withdrawn | CD33 | Hydrazone | Calicheamicin |
Adectris | Brentuximab vedotin | Approved | CD30 | Citrulline-valine | MMAE |
T-DM1 | Trastuzumab emtansine | Approved | HER2 | Thioether | Maytansinoid |
One of the difficulties associated with ADC production is the resulting heterogeneous mixtures after chemical conjugation of cytotoxic drugs. In principle, each of the ADC species in the mixture can come with a different pharmacokinetic, therapeutic, and safety profile [53]. Recently, various strategies have been described to generate more homogeneous ADC, both regarding the sites to which the drugs are attached and the number of drugs that are loaded on an Ab. Generally, a drug-to-antibody ratio of about 2–4 is desirable. Coupling fewer drugs per antibody would compromise efficacy of the ADC. On the other hand, a higher drug load may negatively affect stability, circulation half-life, or binding to the target.
One example of a strategy that yields homogeneous ADCs are THIOMABs, in which the drugs are linked to defined, engineered cysteine residues [54]. Other approaches include enzymatic modification by transglutaminase of deglycosylated mAbs [55] or introduction of non-natural amino acids into the mAb constant regions for subsequent chemical modification [56].
11.4.3 Radioimmunoconjugates
Radioimmunoconjugates are a special class of antibody conjugates with enhanced toxicity. The concept of empowering an mAb with cytotoxic radiation is similar to the ADCs described above. However, the unique properties of radioactive decay entail several special features of radioimmunoconjugates.
Unlike cytotoxic drugs, the radionuclide attached to the antibody does not need to be cleaved from the antibody to become activated. Radioactivity does not only affect the target cell, but also affect neighboring antigen-negative cells. This so-called “cross-fire” effect is one of the attractive features of radioimmunotherapy, especially for tumors with heterogenous antigen expression.
Radionuclides which emit α or β− radiation are most suitable for radioimmunotherapy because of their short range in tissue (Table 11.2). The radiolabeled antibody accumulates at the tumor site, and the energy from the emitted radiation is deposited near the tumor cells. Depending on the type of radiation, single cells, small clusters of cells, or small tumors can be efficiently irradiated [57]. Auger electrons are a third type of radiation which is potentially useful to eradicate single cells. However, due to the very short range in tissue, Auger electrons have to be brought close to the nucleus of the cell to introduce enough DNA damage for efficient cytotoxicity [58]. Therapeutic radionuclides in current clinical trials include primarily the β− emitters 90Y, 177Lu, and 131I [59]. The choice of a suitable radionuclide is not only dependent on the range of the radiation in tissue, but also dependent on the radiochemistry, the physical half-life of the radionuclide, and the availability of the radionuclide. While iodination can be achieved by direct modification of tyrosine residues of an Ab, radiometals have to be attached to proteins using a chelating agent. Two radioimmunoconjugates have been approved for the treatment of non-Hodgkin’s lymphoma, both targeting CD20: 131I-tositumomab (Bexxar) and 90Y-ibritumomab (Zevalin).
Table 11.2
Examples of radionuclides suitable for radioimmunotherapy
Radionuclide | Half-life | Radiation | Maximum range in tissue |
|---|---|---|---|
177Lu | 6.6 days | β− | 2 mm |
90Y | 2.7 days | β− | 12 mm |
131I | 8.0 days | β− | 2 mm
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|